


摘要
胰腺癌是一种侵袭性肿瘤,死亡率接近100%。治疗胰腺癌最成功的药物是吉西他滨,尽管就患者生存而言,其总体效果仍然很差。这项研究是为了评估一种新型的抗胰腺癌的抗癌药物。该组化合物属于双吡啶基硫半氨基脲类,已被证明在体外和体内对一系列不同的肿瘤具有强效和选择性活性。我们首次在胰腺癌中证明,这些药物增加了生长和转移抑制因子N-myc下游调控基因1的表达及其Ser330和Thr346位点的磷酸化,这对其对抗这种肿瘤的活性至关重要。此外,这些药物增加了周期蛋白依赖性激酶抑制剂p21的表达CIP1 / WAF1而cyclin D1在胰腺癌细胞中的表达降低。总之,这些分子的改变部分解释了观察到的明显的抗肿瘤活性。事实上,这些药物在体外抗增殖活性明显高于现有的胰腺癌治疗方法,即吉西他滨和5-氟尿嘧啶。体内研究表明,一种新型硫代氨基脲,即4-环己基-4-甲基-3-硫代氨基脲盐酸盐,能完全抑制胰腺癌异种移植瘤的生长,且正常组织组织学无明显改变。我们的研究共同确定了一种新型和有效的抗肿瘤药物的分子效应,可能对胰腺癌治疗有用。
简介
胰腺癌是一种毁灭性的疾病,98%至100%的病例是致命的,这种疾病的存活率与20年前相同。Jemal等人,2009年).尽管人们越来越多地致力于更好地了解胰腺癌的发病机制和改善胰腺癌的治疗方案(库斯托迪奥等人,2009年;古河道,2009),这种疾病的预后仍然很差。胰腺癌的标准治疗是抗癌药物吉西他滨(图1A),通常与其他化疗药物如5-氟尿嘧啶(图1B) (库斯托迪奥等人,2009年).吉西他滨是核苷脱氧胞苷的类似物,其功能是抑制核糖核苷酸还原酶,并启动DNA链终止和凋亡(Wong等人,2009年).然而,吉西他滨及其与其他药物联合用于胰腺癌治疗的成功是有限的,患者平均寿命仅增加3个月(库斯托迪奥等人,2009年).

吉西他滨(A)、5-氟尿嘧啶(B)、DFO (C)、3-AP (D)、Dp44mT (E)和DpC (F)的化学结构。
考虑到这种疾病的高度侵袭性和开发有效治疗策略的有限进展,我们试图研究一种新的胰腺癌治疗方法,包括靶向生长和转移抑制因子N-myc下游调控基因1 (NDRG1)的产物(Kovacevic和Richardson, 2006;艾伦等人,2008年).后者蛋白在体内抑制胰腺癌的生长、转移和血管生成,从而减少肿瘤的进展(Maruyama等,2006).此外,NDRG1的表达也与胰腺癌分化的增加相关(焦虑等人,2006年).因此,NDRG1可能是治疗本病的一个很有前途的治疗靶点。
在胰腺癌中靶向NDRG1的一种潜在策略是使用新型硫氨基脲类药物,之前已证明通过增加缺氧诱导因子-1 (HIF-1)的能力在体外和体内上调NDRG1 (Le和Richardson, 2004;Whitnall等人,2006年;Kovacevic等人,2008).这种作用的机制是通过硫氨基脲和其他铁螯合剂结合细胞内铁介导的,从而抑制HIF-1α的降解(Le和Richardson, 2004).
铁是多种重要代谢过程所必需的基本元素,包括核糖核酸还原酶,它催化DNA合成中的限速步骤(Kalinowski和Richardson, 2005).铁螯合剂作为抗癌剂的适宜性首次被发现是在铁螯合剂去铁氧胺(DFO;图1C),主要用于治疗铁负荷过重的疾病,如β-地中海贫血(Aouad等人,2002年),成功用于神经母细胞瘤的临床试验(Buss et al., 2003).从那时起,专门为治疗癌症而设计的铁螯合剂已经开发出3-氨基吡啶-2-羧醛硫代氨基吡啶(3-AP;图1D),进入各种I期和II期临床试验(兰德里等人,2010).然而,后者已显示出相当大的问题,包括低疗效和严重的副作用,包括高铁血红蛋白血症和缺氧(Kalinowski和Richardson, 2005).
硫氨基脲可以结合铁和铜,导致氧化还原活性复合物的形成,产生活性氧(ROS),诱导癌细胞的细胞毒性(袁等,2004;Kalinowski和Richardson, 2005;Jansson等人,2010).迄今为止开发的最活跃的硫氨基脲之一是二-2-吡啶基酮4,4-二甲基-3-硫氨基脲(Dp44mT;图1E) (袁等,2004;Kalinowski和Richardson, 2005;Whitnall等人,2006年).Dp44mT已被证明在体外和体内显著降低多种肿瘤的生长,比3-AP更有效,毒性更小(Whitnall等人,2006年).然而,使用高剂量、非最佳剂量的Dp44mT的研究发现,它在裸鼠中诱导心脏毒性(Whitnall等人,2006年).因此,为了开发高效而无毒的硫氨基脲,对Dp44mT进行了修饰,生成了一种新的第二代硫氨基脲,二-2-吡啶基酮4-环己基-4-甲基-3-硫氨基脲盐酸盐(DpC;图1F)。
本研究的目的是研究Dp44mT及其新的类似物DpC的作用机制和活性,在体外和体内。我们证明了这些硫氨基脲影响多种分子靶标,包括NDRG1, p21CIP1 / WAF1在四种胰腺癌细胞类型中的三种中,与目前选择的药物吉西他滨相比,它们在体外抑制增殖和诱导凋亡方面明显更有效。此外,体内研究表明,DpC完全抑制胰腺肿瘤的生长,明显比Dp44mT更有效,毒性更小。因此,DpC可能是一种有效的治疗胰腺癌的新策略。
材料与方法
细胞培养。
胰腺癌细胞系MIAPaCa-2, PANC-1, CAPAN-2和CFPAC-1来自美国类型培养收藏(Manassas, VA)。MIAPaCa-2和PANC-1细胞都是来源于胰腺癌的上皮细胞。CAPAN-2细胞是来源于胰腺腺癌的多边形细胞,而CFPAC-1细胞是来源于胰腺腺癌肝转移的上皮细胞。
MIAPaCa-2、PANC-1和CFPAC-1细胞类型在Dulbecco改良的Eagle's培养基(Invitrogen,悉尼,澳大利亚)中培养,而CAPAN-2细胞在McCoy's培养基(Invitrogen)中培养。所有培养基均添加10% (v/v)胎牛血清,1% (v/v)非必需氨基酸,1% (v/v)丙酮酸钠,2 mMl-谷氨酰胺、100 μg/ml链霉素、100 U/ml青霉素(均来自Invitrogen)。细胞在37°C的恒温箱(Thermo Fisher Scientific, Waltham, MA)中在5% CO的潮湿气氛中生长295%空气和标准方法传代培养,如前所述(Le和Richardson, 2004).
试剂。
吉西他滨(Gemzar)购自礼来公司(Indianapolis, IN)。5-氟尿嘧啶从Sigma-Aldrich(圣路易斯,密苏里州)获得。DFO来自诺华(瑞士巴塞尔)。铁螯合剂Dp44mT的合成和表征如前所述(理查德森等人,2006年).
采用多种方法合成了新型铁螯合剂DpC (斯科维尔,1990;理查德森等人,2006年).简而言之,二硫化碳(0.2 mol)滴入N-甲基环己胺(0.2 mol)在NaOH溶液(250 ml, 0.8 M)中反应,直到有机层几乎消失。接下来,将氯乙酸钠(0.2 mol)添加到水提取物中,并在室温下反应一夜。加入浓盐酸(25 ml)可得到固体羧甲基硫代氨基甲酸酯中间体。然后,将0.08 mol的后一种化合物溶解在20 ml水合肼和10 ml水中。接下来是五个温和加热(直到冒烟)和冷却的循环。然后让溶液静置,直到形成细白色的硫氨基脲晶体。将硫氨基脲(10mmol)水溶液(15ml)加入溶解在乙醇(15ml)中的二-2-吡啶酮(10mmol)中。接下来,加入5滴冰醋酸,将混合物回流2小时,冷却至5℃,得到黄色的Dp4cycH4mT沉淀。最后,将Dp4cycH4mT溶解在最小体积的冰正己烷中,加入等摩尔的HCl,生成HCl盐Dp4cycH4mT·HCl (DpC)。化合物的纯度用元素分析的组合来表征(计算:C, 47.52%; H, 6.82%; N, 14.58%; Found: C, 47.04%; H, 6.54%; N, 15.02%; Department of Chemistry and Biomolecular Sciences, Macquarie University, Sydney, NSW, Australia), infrared spectroscopy, mass spectroscopy, and1H核磁共振波谱(数据未显示)。
Western Blot分析。
如前所述进行蛋白质分离(Dunn等人,2006),而西方分析则通过既定的协议(高和理查德森,2001).所使用的主要抗体是山羊抗人NDRG1 (Abcam Inc., Cambridge, MA);兔抗人p21CIP1 / WAF1、兔抗人pNDRG1 (Ser330)、兔抗人pNDRG1 (Thr346)、兔抗人cleaved PARP、兔抗人Bax、兔抗人Bcl-2(均来自MA Danvers Cell Signaling Technology);小鼠抗人周期蛋白D1和β-肌动蛋白(均来自加州圣克鲁斯生物技术公司)。
流式细胞术。
使用Annexin V和碘化丙啶(PI)标记的流式细胞仪使用标准方法检测硫氨基脲和吉西他滨对细胞凋亡的反应(袁等,2004).简而言之,细胞在T25烧瓶中播种,并让其粘附一夜。然后用10 μM或20 μM吉西他滨、Dp44mT或DpC处理细胞,在37℃下孵育48 h。按照制造商的说明,使用Annexin V细胞凋亡试剂盒(BD Biosciences, San Jose, CA)收集和制备细胞,并使用FACSCalibur流式细胞仪(BD Biosciences)进行检测。结果使用CellQuest软件(BD Biosciences)进行分析。
MTT细胞增殖试验。
细胞增殖用3-(4,5-二甲基噻唑-2-基)-2,5-二苯基四唑(MTT;在72小时/37°C孵育后,使用标准方法(理查德森等人,1995年).如前所述,MTT颜色的形成与活细胞的数量成正比(理查德森等人,1995年),证实了其在这些研究中的使用。
裸鼠最大耐受剂量的研究。
活体实验由动物伦理委员会(悉尼大学)批准。在评估新型硫氨基脲DpC抗肿瘤活性的研究开始之前,进行了最大耐受剂量(MTD)实验,如前所述(袁等,2004;Whitnall等人,2006年)使用BALBc nu/nu裸鼠(动物资源设施,珀斯,西澳大利亚)。MTD定义为30%的队列患者因健康状况明显恶化或体重下降超过10%而死亡的剂量(袁等,2004;Whitnall等人,2006年).
裸鼠肿瘤移植。
在这些研究中,使用了8周大的雌性裸鼠(BALBc nu/nu),并采用标准技术(Whitnall等人,2006年).简言之,每只小鼠皮下注射2 × 106PANC-1细胞悬浮在Matrigel (BD Biosciences)。用游标卡尺测量肿瘤大小,按前文所述计算肿瘤体积(巴尔萨里等人,2004年).一旦肿瘤达到平均90毫米3.,开始治疗(第0天;图7A).螯合剂Dp44mT和DpC溶于30%丙二醇和0.9%生理盐水中,静脉注射(通过尾静脉),每周5天(周一至周五)(Whitnall等人,2006年).吉西他滨溶解于15%丙二醇/0.9%生理盐水中,按照既定方案每3天腹腔注射一次(Laquente等人,2008年).每组小鼠(n= 8)分别接受吉西他滨(5mg /kg)、Dp44mT (0.4 mg/kg)、DpC (5mg /kg)或对照。该治疗方案的实施是基于我们实验室进行的MTD研究和以前使用这些药物的研究(Whitnall等人,2006年;Laquente等人,2008年).车辆对照组被细分为两组(n= 4)第一组静脉注射30%丙二醇/0.9%生理盐水,5天/周,作为铁螯合剂治疗组对照。第二对照组每3天腹腔注射15%丙二醇/0.9%生理盐水,为吉西他滨治疗的适当对照组。一旦对照肿瘤达到1000毫米3.在美国,由于道德要求,这些动物被安乐死。
血液学和组织学。
在体内实验完成后,采用心脏穿刺采血,并采用标准方法测定血液学指标(Dunn等人,2006).包括器官和肿瘤在内的组织被包裹在石蜡块中并进行切片。使用了三种不同的染色剂,即苏木精和伊红,珀尔斯或戈莫里-三色红。组织学分析和病理特征的量化由独立的兽医病理学家Terrence Rothwell博士(Rothwell咨询公司,Avalon Beach, NSW, Australia)进行。
统计分析。
数据比较使用学生的t测试。除非另有说明,结果以均数±标准差表示。当数据被认为具有统计学意义时p< 0.05。
结果
新型硫氨基脲类药物治疗胰腺癌的体外分析。
为了评估新型硫氨基脲对胰腺癌的疗效,并将其与吉西他滨的活性进行比较,我们首先进行了体外研究,检查了关键的分子靶点。其中包括生长和转移抑制因子NDRG1 (Kovacevic和Richardson, 2006),周期蛋白依赖性激酶抑制剂,p21CIP1 / WAF1(Yu等人,2007),以及细胞周期进程所必需的细胞周期蛋白D1 (Yu等人,2007).此外,我们还研究了一些凋亡标志物,以及这些药物诱导细胞凋亡的能力。我们还检测了DFO、Dp44mT和DpC对四种不同类型胰腺癌细胞的体外抗增殖活性,并与该疾病的标准化疗药物吉西他滨和5-氟尿嘧啶(库斯托迪奥等人,2009年).
硫氨基脲类上调胰腺癌细胞生长转移抑制因子NDRG1
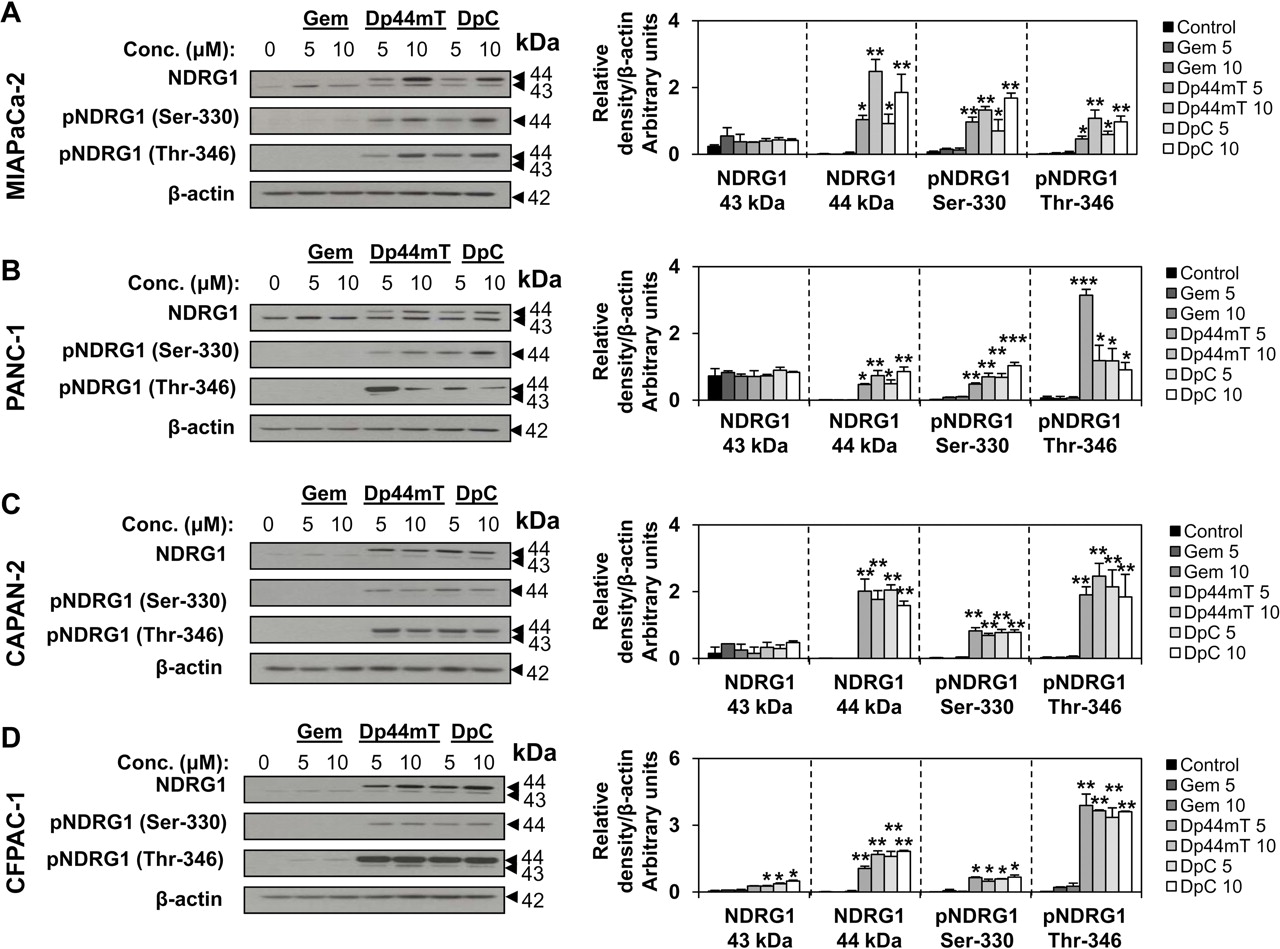
在当前的研究中,我们检测了新型硫氨基脲、Dp44mT和DpC以及吉西他滨对四种胰腺癌细胞类型中总表达和磷酸化NDRG1 (Ser330和Thr346)的影响(图2模拟)。考虑到广泛报道的NDRG1的抗肿瘤功能,这是至关重要的评估(Kovacevic和Richardson, 2006;艾伦等人,2008年)及其作为胰腺癌治疗靶点的潜力(焦虑等人,2006年;Maruyama等,2006).将4种胰腺肿瘤细胞miapca -2、PANC-1、CAPAN-2和CFPAC-1分别用5或10 μM Dp44mT、DpC或吉西他滨孵育24 h/37℃,检测NDRG1蛋白表达。我们的研究结果表明,Dp44mT和DpC均显著(p< 0.05) NDRG1总蛋白和磷酸化(Ser330和Thr346)蛋白水平在所有细胞类型中均上调(图2模拟)。另一方面,吉西他滨没有显著(p> 0.05)与未处理对照组相比,改变了任何评估细胞类型中NDRG1的总表达或磷酸化表达(图2).

硫氨基脲酮Dp44mT和DpC显著上调NDRG1及其Ser330和Thr346位点的磷酸化,而吉西他滨(Gem)则没有作用。将miapca -2 (A)、PANC-1 (B)、CAPAN-2 (C)、CFPAC-1 (D)细胞分别置于含Gem(5或10 μM)、Dp44mT(5或10 μM)、DpC(5或10 μM)的对照培养基或对照培养基中孵育24 h/37℃,Western blotting检测NDRG1表达。在大约43和44 kDa处检测到NDRG1的两个波段,并用密度法定量。针对NDRG1在Ser330和Thr346位点磷酸化的特异性抗体也被用于评估药物对其磷酸化的影响。A、B、C和D中的凝胶照片代表了进行的三个实验,而密度分析为平均值±S.D.(三个实验)。为进行统计学分析,各处理均与未处理对照进行比较;*,p与对照组相比< 0.05;**p与对照组相比< 0.01;***,p与对照组相比< 0.001。
如前所述(默里等人,2004年;Kovacevic等人,2011),我们在每一种被检测的细胞类型中观察到两个总NDRG1条带(迁移在43和44 kDa;图2),这些可能代表了该蛋白的不同磷酸化状态,如前所述(默里等人,2004年).考虑到Dp44mT和DpC均显著(p< 0.05)上调了NDRG1的顶部带,以及该蛋白的两种不同磷酸化形式(Ser330和Thr346;在44kda)中检测的每种细胞类型(图2),上面的条带可能对应于其磷酸化的形式。值得注意的是,当我们用抗体探测Thr346磷酸化的NDRG1时,我们还观察到一个非常微弱的低带,这可能表明另一种NDRG1亚型。虽然NDRG1不同磷酸化状态的生物学相关性尚未得到最终确定,但最近的一项研究表明,NDRG1的磷酸化对其在胰腺癌中的抗肿瘤功能很重要(村上等,2010).因此,我们目前的结果对于了解这些硫氨基脲类的抗肿瘤活性很重要。
新型硫氨基脲调节参与细胞周期进程的其他关键蛋白,即p21CIP1 / WAF1Cyclin D1。
我们最近发现NDRG1可以上调周期蛋白依赖性激酶抑制剂p21的表达CIP1 / WAF1在不同类型的癌细胞中(Kovacevic等人,2011).考虑到这一点,加上p21CIP1 / WAF1由细胞铁含量调节(傅和理查德森,2007),并在预防G1/S进展(Yu等人,2007),我们进一步检查了我们的新硫氨基脲和吉西他滨对p21的影响CIP1 / WAF1表达式。此外,我们还研究了另一种参与细胞周期进展的关键蛋白,即细胞周期蛋白D1的表达,该蛋白已被证明在癌细胞中被铁螯合剂显著降低(Nurtjahja-Tjendraputra等人,2007),是硫氨基脲的另一个潜在分子靶点。
MIAPaCa-2、PANC-1、CFPAC-1和CAPAN-2细胞分别与吉西他滨、Dp44mT或DpC在浓度为5或10 μM的条件下孵育24 h/37°C,并检测p21蛋白水平CIP1 / WAF1检测细胞周期蛋白D1。Dp44mT和DpC均显著(p< 0.05) p21升高CIP1 / WAF1表达,而显著地(p< 0.05)降低了四种检测细胞类型中的cyclin D1水平(图3模拟)。值得注意的是,吉西他滨还能够显著降低MIAPaCa-2、PANC-1和CFPAC-1细胞中的周期蛋白D1水平,而在CAPAN-2细胞中没有显著影响(图3C).另一方面,吉西他滨降低了p21CIP1 / WAF1在PANC-1、CFPAC-1和CAPAN-2细胞中表达,而在MIAPaCa-2细胞中没有观察到相对于对照的影响(图3).因此,吉西他滨的分子效应似乎与细胞类型有关。

吉西他滨(Gem)、Dp44mT、DpC对p21的影响CIP1 / WAF1cyclin D1在胰腺癌细胞中的表达。硫氨基脲酮Dp44mT和DpC显著上调p21CIP1 / WAF1同时显著降低cyclin D1水平。MIAPaCa-2 (A), panan -1 (B), CAPAN-2 (C),和CFPAC-1 (D)细胞。细胞分别在对照培养基或含有Gem(5或10 μM)、Dp44mT(5或10 μM)或DpC(5或10 μM)的培养基中孵育24 h/37°C,然后进行Western blotting。凝胶照片代表了三个实验,而密度分析是平均值±S.D.(三个实验)。为进行统计学分析,各处理均与未处理对照进行比较;*,p与对照组相比< 0.05;**p与对照组相比< 0.01。
综上所述,上述结果表明NDRG1和p21CIP1 / WAF1Dp44mT和DpC显著上调,而cyclin D1在胰腺癌细胞类型中降低。考虑NDRG1在胰腺癌中的抗肿瘤作用(Maruyama等,2006),这些结果表明硫氨基脲类药物可能是对抗这种疾病的一种有益的治疗策略。因此,进一步的体外研究检查了这些药物的抗增殖功效。
与吉西他滨和5-氟尿嘧啶相比,新型硫氨基脲类在体外抑制胰腺癌细胞增殖方面明显更有效。
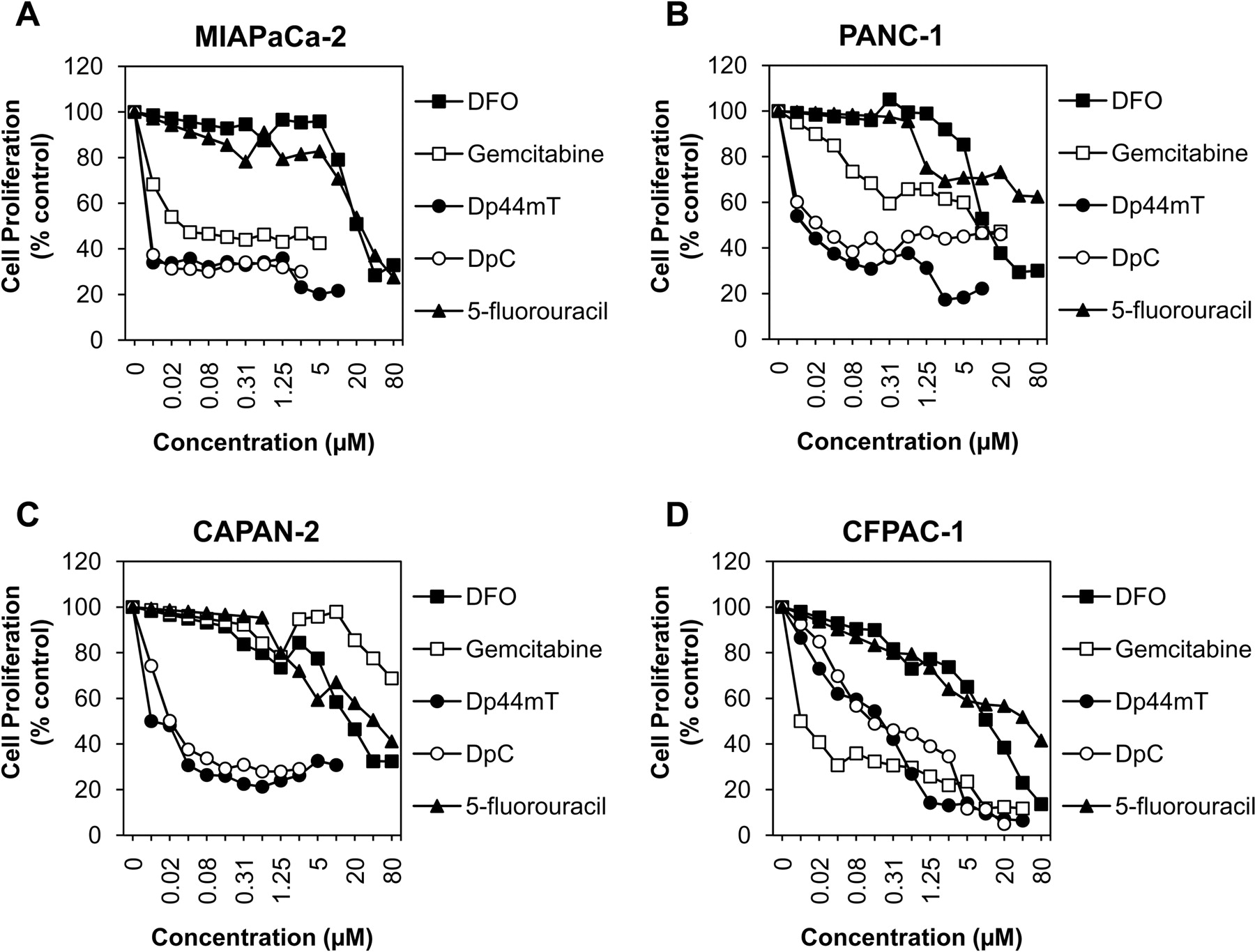
为了检验Dp44mT和DpC对胰腺癌的体外抗增殖活性,我们对上述研究的四种胰腺癌细胞类型分别进行了MTT增殖实验,并与目前使用的治疗该疾病的药物吉西他滨和5-氟尿嘧啶(库斯托迪奥等人,2009年).此外,作为进一步的对照,我们还检查了表征良好的铁螯合剂DFO (Kalinowski和Richardson, 2005).
通过检测MIAPaCa-2、PANC-1和CAPAN-2细胞类型,发现Dp44mT和DpC的抗增殖活性最高(图4, A-C)及其IC50数值显著(p< 0.01)低于吉西他滨和5-氟尿嘧啶(表1).事实上,IC50与吉西他滨和5-氟尿嘧啶相比,四种细胞类型中有三种的Dp44mT和DpC值分别至少低4倍和2000倍(表1).另一方面,DFO的抗增殖活性相对较低,显著(p< 0.001)不如硫氨基脲有效,可能是因为它的膜渗透性低(Kalinowski和Richardson, 2005).

与吉西他滨和5-氟尿嘧啶相比,硫氨基脲酮Dp44mT和DpC抑制胰腺癌细胞增殖更有效。与吉西他滨和5-氟尿嘧啶相比,miapca -2 (A), PANC-1 (B)和CAPAN-2 (C)细胞在37°C孵育72 h后,对Dp44mT和DpC的抗增殖作用更敏感。D, CFPAC-1细胞对低剂量吉西他滨最敏感(< 0.16 μM),对高剂量Dp44mT最敏感(>0.63 μM)。MTT分析如下所述材料与方法.所提供的数据是三到五次实验的平均值,以及计算出的IC50和集成电路90从这些研究中得到的值载于表1和补充表1。
与硫氨基脲类具有最高抗增殖活性的其他细胞类型相比,CFPAC-1细胞对吉西他滨比Dp44mT或DpC更敏感(图4D)事实上,IC50吉西他滨的价值显著(p< 0.05)低于Dp44mT或DpC (图4D;表1).但值得注意的是,Dp44mT和DpC IC较低90比吉西他滨在CFPAC-1细胞中的作用(图4D;补充表1),表明在较高浓度下使用硫氨基脲可有效抑制该细胞类型的体外增殖(图4D).这些结果证明了胰腺癌的异质性,并可能反映了决定对抗癌药物敏感性的不同分子改变(古河道,2009).
吉西他滨与新型硫氨基脲类Dp44mT和DpC均可诱导胰腺癌细胞凋亡。
考虑到Dp44mT和DpC显著改变了许多在生长和转移中起关键作用的蛋白的表达,即NDRG1, p21CIP1 / WAF1,和cyclin D1,我们进一步检查了这些药物的影响,通过评估四种细胞类型的凋亡与吉西他滨比较。
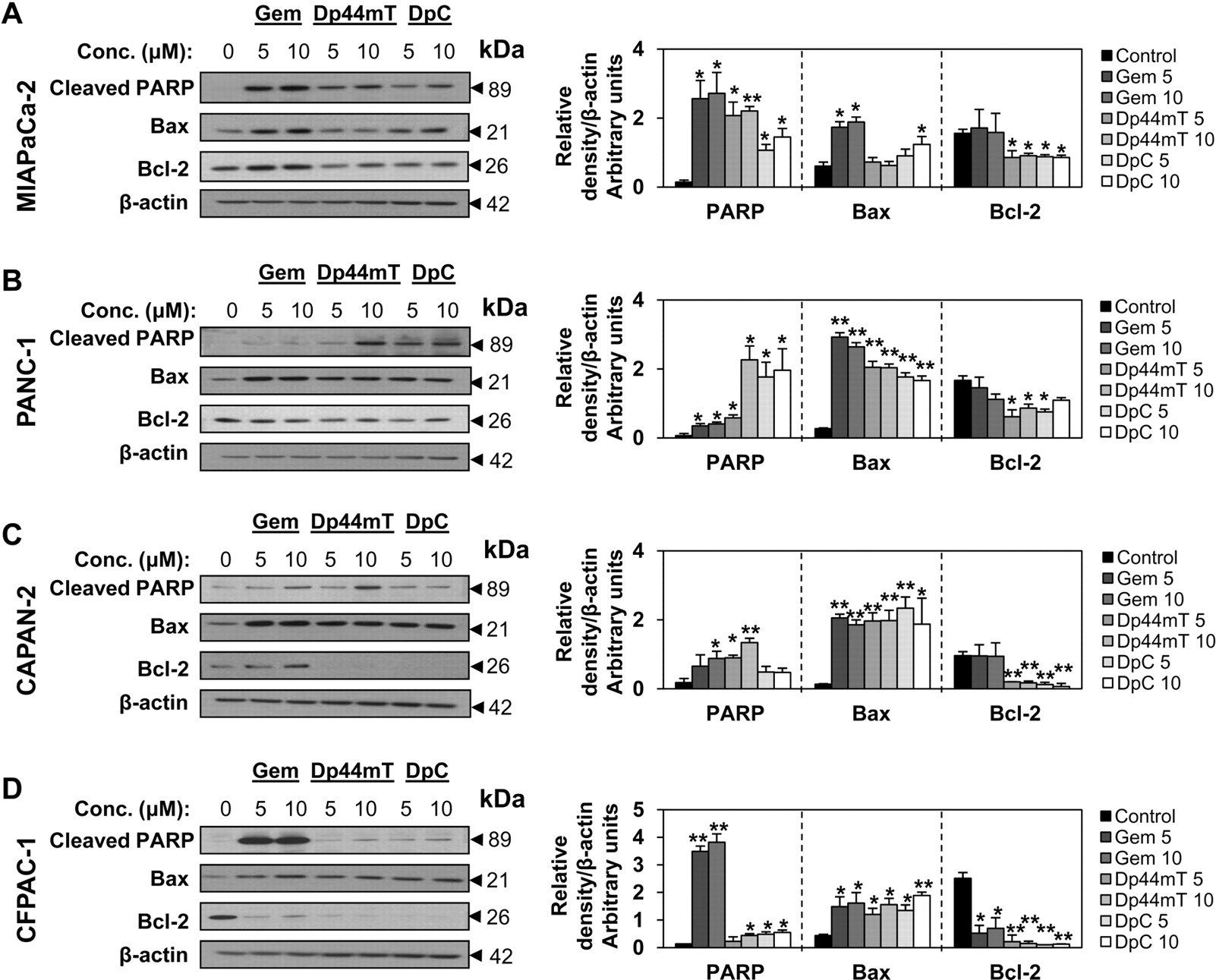
每种细胞类型分别用浓度为5或10 μM的吉西他滨、Dp44mT或DpC孵育24小时,并评估一些凋亡标志物,包括cleaved PARP、Bax和Bcl-2 (唐和波特,1996年).我们观察到,与其他胰腺癌细胞相比,吉西他滨在MIAPaCa-2和CFPAC-1细胞类型中最有效地上调了裂解PARP (图5),这与这些细胞对吉西他滨的敏感性较高相对应(图4而且表1).然而,在这些类型的细胞中,PARP也被硫氨基脲类药物裂解,但程度低于吉西他滨(图5值得注意的是,与吉西他滨相比,panan -1和CAPAN-2细胞对硫氨基脲更敏感或相似,其中Dp44mT (10 μM)对诱导裂解PARP最有效(图5B和C)。

Dp44mT、DpC和吉西他滨调节胰腺癌细胞凋亡的分子标志物。为了研究Dp44mT、DpC或吉西他滨诱导凋亡的能力,我们评估了以下胰腺癌细胞类型:miapca -2 (A)、PANC-1 (B)、CAPAN-2 (C)和CFPAC-1 (D)。细胞与吉西他滨(Gem)、Dp44mT或DpC(5或10 μM)孵育24 h/37°C,然后用Western分析检测裂解的PARP、Bax和Bcl-2。凝胶照片代表三个实验,而密度分析是平均值±S.D.(三个实验)。为进行统计学分析,各处理均与未处理对照进行比较;*,p与对照组相比< 0.05;**p与对照组相比< 0.01。
促凋亡细胞Bax的表达(唐和波特,1996年)在所有四种细胞类型中均被每种化合物上调,但MIAPaCa-2细胞中的Dp44mT除外,在该细胞中未观察到显著影响(图5此外,抗凋亡蛋白Bcl-2的表达(唐和波特,1996年)显著(p< 0.01),而吉西他滨只显著降低了CFPAC-1细胞中的Bcl-2 (图5).进一步的研究检测了这些分子在与这些药物孵育48小时后的表达,结果与24小时后的结果大致相似(补充图1)。上述结果强调了胰腺癌细胞对所检查的药物的不同敏感性,显示了诱导凋亡的各种分子反应。为了阐明细胞凋亡是否发生,以及发生的程度,我们在这些药物孵育后,用流式细胞术进一步检测了胰腺癌细胞。
利用Annexin V和PI标记,我们用10或20 μM浓度的吉西他滨、Dp44mT或DpC处理48 h/37°C后,通过流式细胞仪检测每种细胞的凋亡情况。之所以使用这些浓度,是因为我们的分子研究表明,硫氨基脲在10 μM时诱导细胞凋亡的效果比在5 μM时更好(图5).此外,我们还检测了浓度较高的20 μM,以更好地区分不同药物的凋亡作用。在这些研究中,与相同浓度的吉西他滨和Dp44mT相比,在20 μM浓度下,DpC始终是四种细胞类型中诱导晚期凋亡(Annexin V-和pi阳性)最有效的药物(图6).DpC的作用在MIAPaCa-2、PANC-1和CAPAN-2细胞类型中最为明显,其中DpC的作用显著(p< 0.05)优于同浓度吉西他滨(图6a - c)。然而,在CFPAC-1细胞中,DpC和吉西他滨在两种浓度下均无显著差异,两种药物在该细胞类型中诱导凋亡的效果相同(图6D)。这些结果再次证实了我们早期的发现,即CFPAC-1细胞与所检查的其他细胞类型相比,对吉西他滨更敏感(图4D)。值得注意的是,PANC-1细胞仅对浓度为20 μM的DpC敏感,其他处理对该类型细胞的凋亡无显著影响(图6B)。

DpC在流式细胞术检测的四种胰腺癌细胞类型中都能最有效地诱导晚期凋亡。将miapca -2 (A)、PANC-1 (B)、CAPAN-2 (C)和CFPAC-1 (D)分别用10或20 μM浓度的吉西他滨(Gem)、Dp44mT或DpC孵育48 h/37°C,采用PI和Annexin V (AV)染色流式细胞仪检测细胞凋亡。早期凋亡的细胞数量定义为仅AV阳性的细胞,而晚期凋亡定义为AV和PI均阳性的细胞。以DpC (20 μM)处理的细胞凋亡后期细胞数量最多。所提供的数据代表了进行的三个独立实验,并以平均值±S.D. *表示,p与对照组相比< 0.05;**p与对照组相比< 0.01;***,p与对照组相比< 0.001。DpC处理也与相同浓度的Gem处理进行了比较,用#表示统计学显著性,p< 0.05, ##,p< 0.01。
总的来说,我们的研究结果表明,每一种被检测的药物都能够调节胰腺癌细胞凋亡的标志物。然而,DpC是唯一在所有细胞类型中诱导凋亡的药物,在四种细胞类型中的三种中明显比吉西他滨更有效。
新型硫氨基脲与吉西他滨对胰腺癌的体内研究。
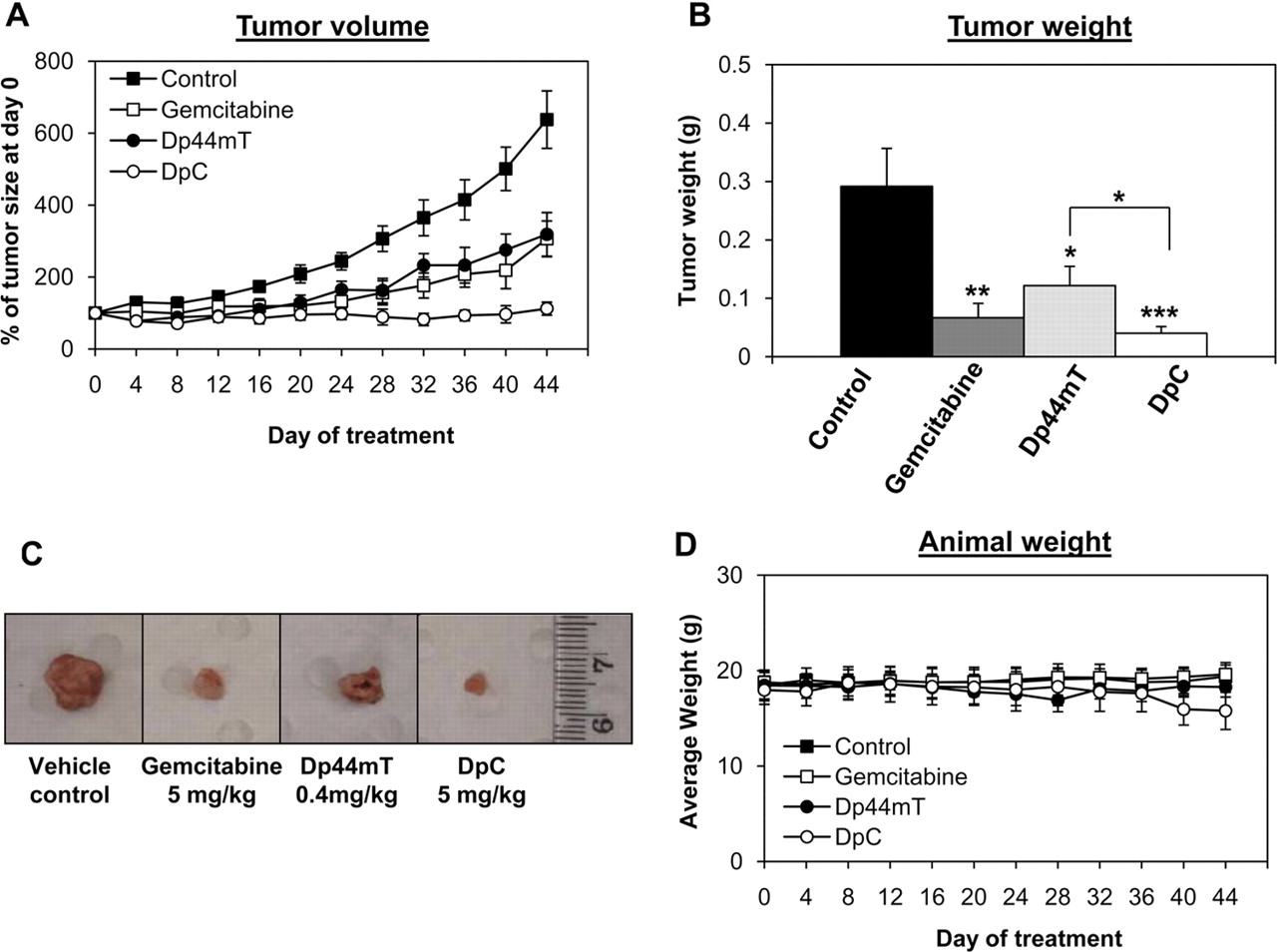
为了进一步表征硫氨基脲类药物对胰腺癌的疗效及其作为一种新型治疗策略的潜力,研究人员在体内对这些药物进行了进一步的研究。在这些实验中,使用了PANC-1细胞,因为它们已被证明适合于产生异种移植,与其他体内胰腺肿瘤相比,这些移植对吉西他滨更耐药(Réjiba等,2009).一旦在裸鼠中建立,肿瘤就可以生长到90毫米3.,然后开始使用单独的载体,吉西他滨(每3天静脉注射5mg /kg), Dp44mT (0.4 mg/kg静脉注射5天/周)或DpC (5mg /kg静脉注射5天/周)治疗。之所以采用这种给药方案和给药途径,是因为在以往的研究中,Dp44mT对其他肿瘤类型表现出良好的耐受性和较高的抗肿瘤疗效(Whitnall等人,2006年),而对于DpC和吉西他滨,初步的最大耐受剂量研究(数据未显示)表明,该给药方案也具有良好的耐受性,并表现出对肿瘤的实质性疗效。
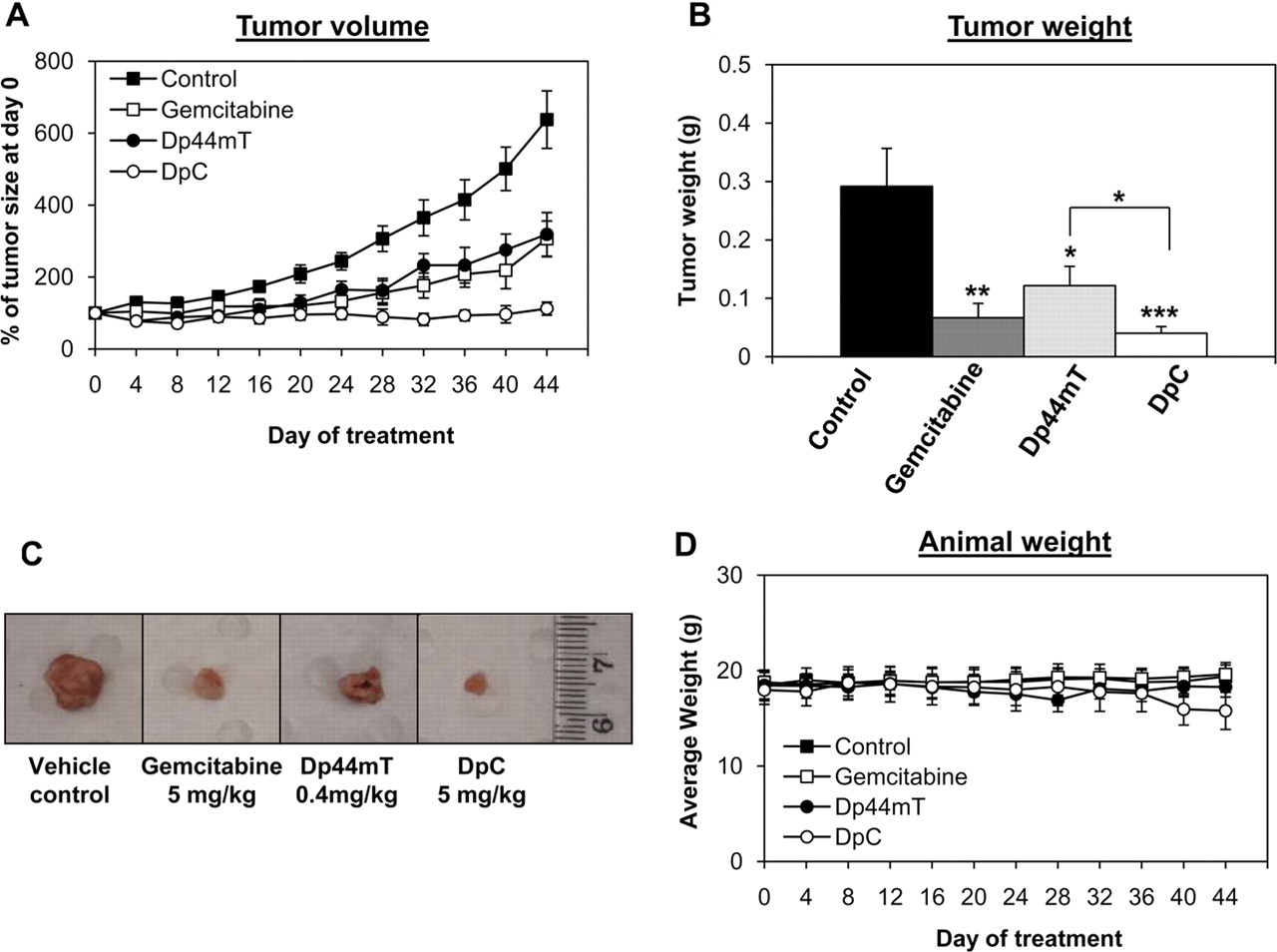
治疗44天后,对照小鼠的平均体积达到675±138 mm3.(图7A)。需要注意的是,由于活性药物是通过这些途径给药的,所以有两组载药对照,分别是腹腔内或静脉内给药(见材料与方法).然而,两个对照组都得到了相同的反应,因此,这些数据一直被组合在一起并作为一个组呈现。用吉西他滨、Dp44mT或DpC治疗小鼠,肿瘤体积减小到202±70、230±52和86±20 mm3.,分别。事实上,吉西他滨(p< 0.01), Dp44mT (p< 0.05), DpC (p< 0.001)均显著降低肿瘤体积,分别为对照组的30±10、34±8和13±3% (图7A).此外,治疗44天后的最终肿瘤重量反映了肿瘤体积。事实上,对照组的肿瘤重量为292±65 mg,而吉西他滨、Dp44mT和DpC治疗的肿瘤明显更小,重量为67±25 (p< 0.01), 122±33 (p< 0.05), 40±12 mg (p< 0.001),分别为(图7B).值得注意的是,DpC显著(p< 0.05)在减轻肿瘤重量方面优于Dp44mT。因此,每种治疗都能显著抑制体内胰腺肿瘤异种移植的生长和进展,其中DpC表现出最大的抗肿瘤疗效(图7a - c)。

Dp44mT、DpC和吉西他滨在体内抑制胰腺癌生长。允许PANC-1肿瘤异种移植生长至90mm3.然后分别使用单药(对照组)、吉西他滨(5 mg/kg i.p.,每3天一次)、Dp44mT (0.4 mg/kg/天i.p., 5天/周)或DpC (5 mg/kg/天i.p., 5天/周)开始治疗。A,各药物均能有效抑制PANC-1胰腺癌异种移植体内生长,DpC完全抑制肿瘤生长。B, DpC和吉西他滨(Gem)治疗动物的平均肿瘤重量最低。DpC显著(p< 0.05)在治疗44天后较Dp44mT更有效地减轻肿瘤重量。C,治疗44天后,对照组、吉西他滨、Dp44mT和DpC组安乐死时代表性肿瘤的照片。D,在研究过程中,每个治疗组动物的平均体重。A、B和D中所示的数据表示为平均值±S.E.M. (n= 8).统计学分析,将各处理与未处理对照进行比较;*,p与对照组相比< 0.05;**p与对照组相比< 0.01;***,p与对照组相比< 0.001。Dp44mT也与DpC进行比较,如图所示。
虽然DpC与吉西他滨之间的差异不显著(p> 0.05),这些数据表明,在第32天之后,吉西他滨和Dp44mT治疗与DpC相比,抑制肿瘤生长的效果越来越差(图7A).由于伦理原因,考虑到载药对照组的肿瘤大小是本实验时间长短的限制因素,44天后不可能继续进一步治疗。这是由于一些对照动物的肿瘤体积达到了当地动物伦理委员会规定的最大限度。然而,未来的研究将验证吉西他滨和DpC的长期疗效,并将进一步区分这两种抗癌药物对胰腺癌的疗效。
检查体重、血液学指标和组织学以确定毒性。
为了确定上述体内研究中使用的不同药物是否与任何毒性相关,安乐死后对小鼠的血液学指数以及身体和器官重量进行了分析。治疗44天后,除DpC外,各组动物体重均接近前处理体重的100% (图7D;表2).这些动物表现出显著的(p< 0.001)较前处理体重减轻12% (表2).虽然我们发现大多数器官重量没有显著差异(表2在不同治疗组之间,我们观察到DpC组也有显著的(p< 0.01)脾脏比对照组小(即0.08比0.12 g;表2).脾脏组织学分析发现各组小鼠的脾红髓中均含有正常的造血细胞群(图8).

对携带PANC-1胰腺肿瘤异种移植的裸鼠进行安乐死后44天的心脏、脾脏和肝脏进行组织学分析,分别使用载体(对照)、吉西他滨、Dp44mT或DpC。研究按照传说中的描述进行图7.黑色箭头仅表示Dp44mT组心肌纤维化。比例尺,200 μm;原始放大倍率,100倍。组织学评估如下所述材料与方法.所显示的图像代表了每一组的结果。进一步的组织学数据分析见补充表2。
在使用这些药物治疗的动物中检查的另一个关键参数是考虑到使用铁螯合剂(Dp44mT和DpC),与贫血潜在副作用相关的血液学指标。我们发现对照组和不同治疗组之间的红细胞、白细胞或血小板计数没有显著差异(表3).然而,我们确实观察到Dp44mT和DpC组有显著的(p< 0.01)降低血红蛋白水平,轻微但显著(p与对照组相比,网织红细胞计数< 0.05)增加(表3).这可能是这些动物轻微贫血的一个指标。
为了进一步研究不同治疗对器官的潜在毒性作用,通过1)苏木精和伊红(检测一般超微结构病理状况),2)珀尔斯(检测铁的存在)和3)戈莫里-三色罗马(检测纤维化)染色,对脾脏、肾脏、肝脏、心脏、肺、大脑和骨髓进行组织学分析。一名独立的兽医病理学家进行了组织学分析,这些结果见补充表2。8只dp44mt处理的小鼠中有2只显示出肝脏中存在造血细胞的证据。在大约一半的Dp44mT和dpc处理的小鼠中,肝脏中也有一些轻度组织病理状况的证据。此外,在8只对照组小鼠中的4只以及所有Dp44mT和dpc处理的动物的肾脏中发现了铁沉积物(补充表2)。这些观察结果可能分别与饮食中的铁和尿液中螯合铁复合物的排泄有关。另一方面,吉西他滨治疗组肾脏中没有铁沉积的证据(补充表2)。此外,Dp44mT组每只小鼠的心肌均显示心肌病变,其特征为心肌纤维变性和坏死,并被纤维组织替代(图8和补充表2)。观察到的病理变化在右心室壁和左心室心内膜下的心肌最为明显(图8).这与早期的一项研究一致,该研究也在dp44mt处理的裸鼠中检测到心脏纤维化(Whitnall等人,2006年).值得注意的是,dpc治疗组的心脏没有纤维化病变的证据,这表明该化合物在使用剂量下表现出强大的抗肿瘤活性,并且在体内的毒性远低于Dp44mT。值得注意的是,这些结果代表了这类化合物的选择性抗肿瘤活性的实质性改进。在检查的任何其他器官中都没有明显的病理组织(补充表2),这表明与对照治疗组相比,DpC和吉西他滨都没有引起明显的组织损伤。
讨论
胰腺癌是一种侵袭性疾病,目前可用的治疗方法反应不佳,包括标准的吉西他滨(Jemal等人,2009年).为此,我们研究了一类新的硫氨基脲,旨在针对关键的营养铁(理查德森等人,2009年).硫氨基脲已被发现对一系列不同的肿瘤具有有效和选择性的活性(袁等,2004;Kalinowski和Richardson, 2005;Whitnall等人,2006年).事实上,这些药物也被证明可以克服化疗耐药(Whitnall等人,2006年),这在胰腺癌的治疗中是一个明显的问题(库斯托迪奥等人,2009年).然而,这些新型硫氨基脲类药物对胰腺癌的疗效尚未得到评估。
硫氨基脲和其他铁螯合剂可能是治疗胰腺癌的合适策略的第一个指标之一是发现它们上调了一系列癌细胞类型的生长和转移抑制因子NDRG1 (Le和Richardson, 2004;Whitnall等人,2006年;Kovacevic等人,2008).事实上,早期的研究已经证明,铁的消耗是导致NDRG1水平增加的原因,这部分是通过HIF-1 (Le和Richardson, 2004).与这些研究一致的是,我们发现Dp44mT和DpC在检测的四种胰腺癌细胞类型中都显著增加了NDRG1的表达和磷酸化,而吉西他滨没有显著调节其表达。磷酸化的NDRG1对硫代氨基脲的反应增加在这些药物的作用机制方面具有重要意义,因为NDRG1在Ser330和Thr346位点的磷酸化对于其在胰腺癌中的抗肿瘤活性是必要的(村上等,2010).
我们已经证明NDRG1也能够上调周期蛋白依赖性激酶抑制剂p21CIP1 / WAF1在一些癌细胞类型中(Kovacevic等人,2011).在这里,我们进一步证明了Dp44mT和DpC也增加了p21CIP1 / WAF1与NDRG1水平升高相关。值得注意的是,一项早期检测MCF-7乳腺癌细胞的研究表明,其他铁螯合剂[DFO和2-羟基-1-萘醛异烟碱酰(311)]降低了p21CIP1 / WAF1蛋白质水平(傅和理查德森,2007).这可能表明p21CIP1 / WAF1螯合剂可能是细胞类型特异性的或依赖于所使用的配体类型,因为螯合剂根据其结构表现出不同的效果。事实上,硫氨基脲在结合铁上诱导ROS生成,而DFO和311结合铁而不诱导ROS (Kalinowski和Richardson, 2005).与硫氨基脲类相比,吉西他滨降低了p21CIP1 / WAF1四种胰腺癌细胞类型中三种的表达。
值得注意的是p21的功能CIP1 / WAF1在细胞周期调控中是复杂的,其过表达导致G1由于其作为周期蛋白依赖性激酶抑制剂的能力,/S的阻滞,而p21的减少CIP1 / WAF1表达诱导细胞凋亡(Cheng等,1999).事实上,p21的基础表达CIP1 / WAF1是稳定细胞周期蛋白D1/cdk复合物所必需的,这是细胞周期进程所必需的(Cheng等,1999).因此,p21CIP1 / WAF1与吉西他滨孵育后,在一些细胞中的表达可能通过诱导凋亡来促进其抗肿瘤活性,而p21增加CIP1 / WAF1如本研究所示,对硫氨基脲的反应水平可以抑制细胞周期进程和增殖。
我们还检测了另一个重要的细胞周期调节分子,细胞周期蛋白D1的表达,它与细胞增殖有关(Yu等人,2007).Dp44mT和DpC都降低了每种细胞类型中的周期蛋白D1水平,这与早期的一项研究一致,该研究也证明了铁螯合剂降低周期蛋白D1的能力(Nurtjahja-Tjendraputra等人,2007).吉西他滨还能够显著降低四种细胞类型中的三种细胞的周期蛋白D1水平,正如先前使用胰腺癌细胞所证明的那样(Kunnumakkara等人,2007年),但对其在CAPAN-2细胞中的表达无影响。值得注意的是,cyclin D1在胰腺癌中起致癌基因的作用,通常在这些肿瘤中过度表达,并与患者生存率差相关(Kornmann等人,1998年).因此,能够有效降低cyclin D1水平的抗癌药物可能有利于胰腺癌的治疗。
考虑到硫氨基脲对上述三个关键分子靶点的显著影响,令人感兴趣的是,在抑制四种胰腺癌细胞类型中的三种细胞的增殖方面,Dp44mT和DpC比吉西他滨有效4倍,在所有细胞类型中,>比5-氟尿嘧啶有效2000倍。总的来说,我们的体外分析表明,与吉西他滨和5-氟尿嘧啶相比,新型硫氨基脲酮Dp44mT和DpC更有效地抑制胰腺癌细胞的增殖。因为这些药物上调了NDRG1和p21CIP1 / WAF1这些分子在诱导细胞凋亡中的表达和作用(Stein等人,2004年;Yu等人,2007),我们进一步对比吉西他滨对细胞凋亡的影响。每种药物都能调节细胞凋亡的标志物,包括cleaved PARP, Bax和Bcl-2。然而,这些影响的程度取决于细胞类型和药物浓度。通过流式细胞术,我们证明DpC是诱导晚期凋亡最有效的药物,在四种胰腺癌细胞类型中的三种中明显比吉西他滨更有效。
值得注意的是,CFPAC-1细胞对硫氨基脲的敏感性始终低于其他三种细胞类型,但更容易受到吉西他滨的抗增殖作用的影响。这些结果表明,CFPAC-1细胞具有其他分子属性,使它们对这些药物更具抗性。事实上,最近的研究表明,与PANC-1细胞相比,CFPAC-1和MIAPaCa-2细胞具有更高的内源性ROS水平,这使得它们对吉西他滨更敏感(Donadelli等人,2007年).这与我们的结果一致,在测试的胰腺肿瘤细胞类型中,CFPAC-1和MIAPaCa-2细胞对吉西他滨最敏感。目前尚不清楚内源性ROS水平如何导致CFPAC-1细胞对硫氨基脲的更大抗性。然而,较高的ROS可能导致这些细胞具有增强的抗氧化防御机制(例如,增加过氧化氢酶等),因为这些硫氨基脲的细胞毒性效应机制是由于它们产生ROS的能力(袁等,2004;理查德森等人,2006年),这可能解释了CFPAC-1细胞对这些药物的更强抗性。
在体内研究这些药物对PANC-1胰腺癌异种移植的疗效发现,最有效的治疗方法是DpC,它似乎完全抑制了肿瘤的生长。考虑到DpC对胰腺癌的高疗效,检查这种治疗方案的任何潜在毒副作用是很重要的。与其他治疗组相比,我们注意到DpC治疗的最后一天体重减轻(12%)。此外,DpC-和dp44mt -处理组均表现出网织红细胞计数轻微但显著增加,血红蛋白水平下降。这可能表明轻度贫血,并强调建立一个有效的治疗方案的重要性,将克服这些副作用,同时保持抗肿瘤活性。
本研究的另一个重要结果是两种硫氨基脲类化合物Dp44mT和DpC的比较。早期研究在裸鼠体内检测Dp44mT对抗黑素瘤异种移植,发现在较高剂量的这种螯合剂时,一些心脏纤维化(Whitnall等人,2006年).值得注意的是,我们还观察到Dp44mT (0.4 mg/kg)治疗的小鼠心肌纤维化有限,而DpC (5 mg/kg)治疗后没有心脏毒性。因此,DpC能够克服Dp44mT观察到的主要毒性,同时保持强大的抗癌活性。这些结果清楚地表明DpC是我们实验室开发的最有效和最选择性的铁螯合剂,并值得进一步研究其治疗胰腺癌的潜力。
值得注意的是,硫氨基脲3-AP已通过临床试验,用于治疗一系列肿瘤,包括胰腺癌(Attia等人,2008年).事实上,II期临床试验是使用3-AP联合吉西他滨进行的,因为观察到这些药物具有协同作用(Mackenzie等人,2007年).然而,这些研究发现3-AP引起了显著的毒性,几乎没有治疗益处(Mackenzie等人,2007年;Attia等人,2008年).考虑到Dp44mT比3-AP更有效,毒性更小(袁等,2004;Whitnall等人,2006年), Dp44mT和DpC都是新的、更有效的替代品。因此,研究吉西他滨和DpC之间的潜在协同作用是有必要的,可能会产生更有效的治疗方案。
总之,本研究首次检验了新型硫氨基脲类化合物对胰腺癌的抗癌活性。我们证明了Dp44mT和DpC上调了NDRG1和p21CIP1 / WAF1下调细胞周期蛋白D1,这是抑制细胞增殖的关键分子靶点。此外,DpC通常比目前的标准治疗,即吉西他滨和5-氟尿嘧啶更有效。其他研究表明,DpC完全抑制胰腺肿瘤异种移植物的生长,与Dp44mT不同,DpC不会导致心脏纤维化。这些数据清楚地强调了DpC作为胰腺癌有效治疗策略的潜力。
作者的贡献
参与研究设计:科瓦切维奇,洛夫乔伊和理查德森。
进行实验:科瓦切维奇和奇哈尼。
贡献了新的试剂或分析工具:洛夫乔伊。
数据分析:科瓦切维奇和理查德森。
手稿的:撰写或贡献手稿的:科瓦切维奇和理查德森。
致谢
我们感谢铁代谢和螯合项目(悉尼大学)的Christopher Austin博士、Katie Dixon博士、Patric Jansson博士、Yu Yu博士、Darius Lane博士、Angelica Merlot博士和张道海博士在提交前对手稿的严格评估。
脚注
↵
 本文的在线版本(可在http://molpharm.aspetjournals.org)包含补充材料。
本文的在线版本(可在http://molpharm.aspetjournals.org)包含补充材料。这项工作得到了澳大利亚国家健康和医学研究委员会的支持[Grant 632778;高级首席研究员奖学金571123];新南威尔士州癌症研究所[Grant 06/RSA/1-12];以及澳大利亚扶轮社健康研究基金。
文章、发表日期和引文信息可在http://molpharm.aspetjournals.org.
doi: 10.1124 / mol.111.073627。
缩写:
- NDRG1
- N-myc下游调控基因
- 伯灵顿
- bcl -2相关X蛋白
- bcl - 2
- b细胞CLL/淋巴瘤
- 柴油
- desferrioxamine
- DpC
- 二-2-吡啶酮-4-环己基-4-甲基-3-硫氨基脲盐酸盐
- Dp44mT
- 4-dimethyl-3-thiosemicarbazone di-2-pyridylketone 4日
- HIF-1
- 低氧诱导因子- 1
- MTD
- 最大耐受剂量
- PARP
- 保利(ADP-ribose)聚合酶
- 3-AP
- 3-aminopyridine-2-carboxaldehyde thiosemicarbazone
- 311
- 2-羟基-1-萘醛异烟酰腙
- AV
- 膜联蛋白V
- 麻省理工
- (3) - 4 5-dimethylthiazol-2-yl 2, 5-diphenyl四唑
- π
- propidium碘化
- ROS
- 活性氧。

- 收到了2011年5月18日。
- 接受2011年6月30日。
- 版权所有©2011美国药理学和实验治疗学会








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}