


摘要
为了从体外数据预测肝代谢抑制剂在肝脏中的积累程度,我们研究了分离的大鼠肝细胞中细胞/培养基浓度比(C/M比)和肝/血游离浓度(K男朋友)在静脉注射各种代谢抑制剂后,如伊曲康唑、酮康唑、维拉帕米、地尔硫卓、依诺沙星、环丙沙星、克拉霉素、西咪替丁和尼扎替丁。伊曲康唑在0.1 μg/ml和10 μg/ml浓度下的C/M比分别为~ 6000和200,酮康唑和维拉帕米进入肝细胞也表现出浓度依赖性,但程度小于伊曲康唑。肝细胞对地尔硫卓、依诺沙星、环丙沙星和克拉霉素的吸收呈浓度依赖性线性分布。C/M比值与K之间具有极好的相关性男朋友9种药物的斜率均为1。这一发现提示了预测肝脏中药物浓度的可能性(CH),从C/M比值,药物的血液浓度(CB)和血液中的游离组分(fB),用C表示H= (c / m)·cBf·B.从K可以预测人肝脏中的药物浓度男朋友用与大鼠相似的方法对分离的人肝细胞和血液中的浓度进行估值。
在临床病例中,许多药物-药物相互作用的不良影响已被报道。肝脏和/或肠道中一种药物对另一种药物的代谢抑制是药代动力学药物-药物相互作用中最重要的事件之一(费等人,1987;Olkkola等人,1993年,1994,1996;Backman等人,1994年;Ahonen等人,1995;Baldwin等人,1995).如果使用肝微粒体、原代培养的肝细胞和/或cypa表达的细胞获得代谢抑制常数,我们可以在一定程度上定量估计相互作用的程度(Pichard等人,1990年;Gascon和Dayer, 1991年;Wrighton和Ring, 1994;Ghosal等人,1996年).而浓度-时间曲线下面积(AUC)的预测增长率1用血浆中游离浓度作为体内抑制剂浓度,有时会大大低估相互作用药物的浓度。通过体外代谢抑制实验,定量预测咪达唑仑(MDZ)在抗真菌药物伊曲康唑(ITZ)和酮康唑(KTZ),西咪替丁(CIM)和尼扎替丁(NIZ),组胺H2受体拮抗剂作为抑制剂同时使用(Takedomi等人,1998年;Yamano等人,1999年).用血浆中未结合浓度作为抑制剂在代谢酶附近的浓度,预测值被大大低估,而肝脏中未结合浓度的预测值与观测值非常接近,这表明有必要考虑抑制剂进入肝脏的浓度摄取。在临床情况下,需要通过人类肝脏中抑制剂的浓度来预测其浓度的增加比例;然而,很难直接测量肝脏中的药物浓度。因此,有必要开发一种方法,从血浆中的浓度来估计肝脏中的浓度,血浆的浓度实际上是可以测量的。在本研究中,我们试图通过对大鼠分离肝细胞的体外摄取数据来预测其在肝脏中的浓度。我们检查了细胞/培养基浓度比(C/M比)和肝脏/血液游离浓度(K男朋友)大鼠肝脏值。
实验程序
材料。
ITZ和KTZ由Janssen-Kyowa公司(东京,日本)提供。依诺沙星(ENX)和环丙沙星(CPFX)由大日药厂(大阪,日本)提供。克拉霉素(CAM)由大正制药株式会社(日本东京)供应。盐酸维拉帕米(VER)和盐酸地尔硫卓(DLZ)购自Wako Pure Pharmaceutical Co. (Osaka, Japan)。所有其他作为试剂使用的化学品都是试剂级和高效液相色谱试剂。
动物。
雄性sd大鼠(230 ~ 260克)购自日本东京生物供应中心。老鼠被允许自由地获得水和食物颗粒。
药物溶液的制备。
将ENX和CPFX溶于少量1n NaOH中,用少量0.5 N HCl中和,然后用生理盐水稀释,制备5 mg/ml ENX或CPFX溶液。将VER和DLZ溶于生理盐水中制备5和20 mg/ml VER或DLZ溶液。CAM溶于等摩尔HCl中,用0.1 N NaOH中和,然后用生理盐水稀释,制备5 mg/ml溶液。
肝脏和血浆(血液)浓度和肝脏/血液(游离)浓度比。
在轻乙醚麻醉下,大鼠经股静脉和股动脉插管。麻醉恢复后,经股静脉给药ENX (10 mg/kg)、CPFX (10 mg/kg)、CAM (10 mg/kg)、VER (5 mg/kg)、DLZ (5 mg/kg)。分别于给药后2、5、10、20、30、45、60、90、120、180 min从股动脉采集血液样本,以12000 rpm离心2 min获得血浆。然后在给药180分钟后取出肝脏,血浆和肝脏保存在−20°C,直到分析。肝脏用4卷冰水蒸馏水匀浆。每种药物在血浆和肝脏中的浓度用后面介绍的方法测定,给药后180 min的肝脏/血浆浓度比被视为表观肝脏/血浆浓度比(KP)的值。
用非线性最小二乘法(MULTI)拟合以下双指数方程(Yamaoka等,1981):
此外,对于VER, DLZ, ENX, CPFX,和CAM,真实KP数值是通过修正表观来计算的KP值如下。在轻乙醚麻醉下,大鼠股动脉和门静脉插管。麻醉恢复后,经门静脉注射ENX (10 mg/kg)、CPFX (10 mg/kg)、CAM (10 mg/kg)、VER (20 mg/kg)、DLZ (20 mg/kg)。分别于给药后2、5、10、20、30、45、60、90、120和180 min采集血液样本,并按上述方法测定血浆中的药物浓度。采用非线性最小二乘法(MULTI)计算药代动力学参数(Yamaoka等,1981).根据静脉给药后的AUC (AUC)计算肝提取比(E)注射。)和门静脉内给药后的AUC (AUC光伏)被视为eq。5:
真实肝血浓度比(KB)值根据eq计算。7(Lin等人,1982年):
明显的KP(KP,应用) ITZ、KTZ、CIM和NIZ的值引用于我们的报告(Takedomi等人,1998年;Yamano等人,1999年).ITZ和KTZ的肝廓清量远小于肝血流速率。对于KTZ, CIM和NIZKP,应用当给药后血浆浓度达到稳态(β = 0)时,取KP,应用值(Takedomi等人,1998年;Yamano等人,1999年).因此,KP,应用ITZ、CIM、NIZ、KTZ的值均为实数KP值。
肝脏中真实浓度/血液中游离浓度比值(K男朋友)按公式计算。8:
fP采用平衡透析法评价VER、DLZ、ENX、CPFX和CAM的变化。透析使用透明丙烯酸树脂制成的装置,包括两个1.5 ml的腔室,由纤维素透析膜隔开(SC-101-M10H;Diachema,苏黎世,瑞士)。每种药物分别以5和20 μg/ml的浓度添加到大鼠新鲜血浆中,分别应用于一个腔室,另一个腔室应用等渗磷酸盐缓冲液(pH 7.4)。在37°C孵育6小时后,从两个室中收集0.1 ml样品用于测定。fP按公式计算。9.fP摘录自我们的报告(Takedomi等人,1998年;Yamano等人,1999年).
CB/ CP测量各种药物的比例如下。将每种药物分别以0.5、2、10 μg/ml的浓度加入大鼠新鲜血液中,取1 ml血样,37℃孵育15 min,取0.2 ml血样,离心、CB/ CP计算了比率。从初步实验中,我们证实了CB/ CP在37°C孵育15分钟后,比例基本不变,每种药物在孵育期间是稳定的。
离体大鼠肝细胞摄取动力学。
大鼠肝细胞的分离方法鲍尔等人(1975).细胞活力通过台盼蓝排除试验检测,在85 ~ 95%的范围内。用比色法测定蛋白质浓度洛瑞等人(1951).所有实验均在细胞制备后2 h内完成,此时活性无明显变化。
对分离的大鼠肝细胞吸收各种药物的时间过程进行了如下研究。分离的大鼠肝细胞(蛋白浓度20 mg/ml)悬浮在Krebs-Henseleit缓冲液中。然后,将0.3 ml的肝细胞悬液和2.7 ml的Krebs-Henseleit缓冲液(无白蛋白)混合,在37℃预培养5分钟。在每个肝细胞悬液中以1 μg/ml的浓度加入30微升的各种药物标准溶液,37℃培养。在加药后的20、40、60、120、300 s,去除400 μl细胞悬液。对于ITZ,也在600 s时取样。将细胞悬浮液置于1.5 ml聚乙烯管中,之前分层加入500 μl硅油(比重,1.050)和200 μl 3n KOH。然后,样品在能够极快速加速的台式微fuge中离心10秒,以将细胞从培养基中分离。样品在- 20°C冷冻,然后在油层中间切割样管。测定其在上层(中)和下层(肝细胞)的浓度,以研究其摄取到分离的大鼠肝细胞的时间过程。药物的摄取根据黏附液的体积进行校正,然后转换为真实的细胞内浓度。 The values of adherent fluid (2.2 μl/mg protein) and intracellular space (5.2 μl/mg protein) were obtained from the literature (Miyauchi等,1993).
对分离的大鼠肝细胞摄取药物的浓度依赖性进行了如下研究。ITZ、KTZ、VER、DLZ的药物浓度分别为0.1、0.2、0.5、1、2、5、10 μg/ml, ENX、CPFX、CAM的药物浓度分别为0.1、1、10 μg/ml。对分离的大鼠肝细胞的摄取实验如上所述。每种药物的孵育时间足以达到平衡(KTZ、VER、DLZ、ENX、CPFX和CAM为5分钟;10分钟为ITZ)。
各种药物浓度的测定。
血浆和血液中ITZ和KTZ浓度的测定方法参照既往报道(Yamano等人,1999年).
为了测定血浆、血液和肝脏中VER和DLZ的浓度,取0.1 ml血浆或血液,或0.5 ml 20%的肝脏匀浆与0.1 ml甲醇、0.5 ml 1 N NaOH和2.5 ml异丙醚混合,摇匀5分钟,然后3000转离心5分钟。将2毫升有机相转移到另一管中,在氮气下蒸发。用0.2 ml流动相溶解,注入75 μl高效液相色谱。色谱系统由自动进样器717 (Waters, Tokyo, Japan)、泵LC-10AD和SPD-10A可变波长紫外检测器(Shimadzu Corp., Kyoto, Japan)组成,工作波长分别为229 nm和237 nm,用于VER和DLZ。色谱柱为反相Inertosil ODS, 4.6 mm × 250 mm (GL Science, Osaka, Japan),保持在40°C。流动相为乙腈-10 mM磷酸盐缓冲液(pH 6.5) (80:20, v/v),用于VER,乙腈-10 mM磷酸盐缓冲液(pH 3.0) (35:65, v/v),用于DLZ,以1 ml/min的流速等压泵送。血浆和血液的定量下限为50 ng/ml,肝脏为500 ng/g。
为了在分离的细胞或培养基中测定VER和DLZ,将0.5 ml 1 N NaOH和5 ml异丙醚混合,摇5分钟,然后以3000转/分离心5分钟。然后将4毫升有机相转移到另一个管中,用3 ml 0.1 N HCl反萃取。在2ml水相中加入0.5 ml的1n NaOH,用2.5 ml的异丙醚提取。将两毫升有机相转移到另一管中,在氮气下蒸发。用0.2 ml流动相溶解,注入75 μl高效液相色谱。HPLC条件与血药浓度测定相同。
用改进的方法测定血浆和血液中CIM和NIZ的浓度Takedomi等人(1998).简单地说,0.1 ml血浆或血液,0.1 ml甲醇,0.5 ml 1 N NaOH, 5 ml二氯甲烷混合摇5分钟,然后3000转离心5分钟,然后将4毫升有机相转移到另一管中,在氮气下蒸发。用0.2 ml流动相溶解,注入40 μl高效液相色谱。检测波长为228 nm。色谱柱为反相YMC- pack Pro C18, 3.0 mm × 150 mm (YMC, Kyoto, Japan),保持在40。流动相为乙腈-10 mM磷酸盐缓冲液(pH 6.5, 15:85, v/v),以0.4 ml/min的流速等速泵送。血浆和血液的定量下限均为100 ng/ml。
为了测定血浆中的ENX和CPFX浓度,将0.1 ml血浆、0.2 ml甲醇、1 ml 100 mM磷酸盐缓冲液(pH 7.4)和5 ml含1%氯乙酸乙酯的氯仿混合,摇10分钟,然后以3000转/分离心5分钟。然后将4毫升有机相转移到另一管中,在氮气下蒸发。用0.2 ml流动相溶解,注入75 μl高效液相色谱。HPLC法测定ITZ和KTZ的浓度方法相同。紫外检测器的波长设置为270 nm。色谱柱为反相YMC- pack ODS-H, 4.6 mm × 250 mm (YMC),保持在40。流动相为乙腈-10 mM磷酸盐缓冲液(pH 3.0, 50:50, v/v),以1ml /min的流速等速泵送。血浆中ENX和CPFX的定量下限均为100 ng/ml。
ENX和CPFX浓度的测定血液、肝脏、肝细胞悬液,或中等,0.1毫升的血液或0.5毫升的20%肝匀浆,0.2毫升的甲醇,1毫升的100毫米磷酸盐缓冲剂(pH值7.4),包含10%和5毫升氯仿isopropylalchol混合样品和动摇了10分钟,然后在3000转离心5分钟。4毫升的有机相然后转移到另一个管和反萃取3毫升0.01 N氢氧化钠。在2 ml水相中加入0.4 ml 0.05 N HCl和1 ml 100 mM磷酸盐缓冲液(pH 7.4),用含1%氯乙酸乙酯的5 ml氯仿提取。血液定量下限为100 ng/ml,肝脏定量下限为500 ng/g。
用于测定血浆和肝脏中的CAM浓度,0.1 ml血浆或0.5 ml 20%的肝匀浆,0.5 ml 0.5 N NaOH和3 ml叔-丁基甲基醚混合摇5分钟,3000转离心5分钟,再将2毫升有机相转移到另一管中,在氮气下蒸发。用50 μl流动相溶解,20 μl注入HPLC。色谱系统由一台LC-10AD泵和一台ECD-10A电子化学检测器(岛津公司)组成。氧化的检测细胞电位为1300 mV。色谱柱为反相TSKgel 80TM ODS, 4.6 mm × 150 mm (Toso, Tokyo, Japan),保持在30。流动相为乙腈-100 mM磷酸盐缓冲液(pH 6.4, 50:50, v/v),以1ml /min的流速等速泵送。血浆和血液的定量下限为300 ng/ml,肝脏为1000 ng/g。
在分离细胞或培养基中测定CAM时,用0.5 ml的0.5 N NaOH和5 ml的叔-丁基甲基醚混合并摇晃5分钟,然后以3000转/分离心5分钟。然后将4毫升有机相转移到另一个管中,用3毫升100mm磷酸盐缓冲液(pH 4.0)反萃取。在2ml水相中加入0.5 ml的1n NaOH,用2.5 ml的NaOH提取叔丁methylether。将两毫升有机相转移到另一管中,在氮气下蒸发。用50 μl流动相溶解,20 μl注入HPLC。HPLC条件与血药浓度测定相一致。
在所有测量中,变异系数<10%,运行内精度<±10%。当样品中的浓度低于定量限度时,通过增加样品的量来确定水平。
统计分析。
使用Student 's进行统计分析t测试。差异被认为有统计学意义时p数值<.05。
结果
各种药物的血药浓度和肝脏提取比。
数字1和表1显示静脉和静脉给药后VER、DLZ、ENX、CPFX和CAM的血药浓度分布和药代动力学参数。VER、DLZ、ENX、CPFX和CAM的肝提取比分别为0.797、0.782、0.228、0.529和0.088。

大鼠静脉注射VER、DLZ、ENX、CPFX和CAM后,血药浓度变化曲线(A)、DLZ (B)、ENX (C)、CPFX (D)、CAM (E)。
VER、DLZ、ENX、CPFX和CAM分别以5、5、10、10和10 mg/kg的剂量经股静脉给药。分别以20、20、10、10、10 mg/kg的剂量经门静脉给药VER、DLZ、ENX、CPFX和CAM。每一个点和竖线代表平均值±标准差(n= 4 - 7)。
CB/ CP比率,KBValues和fP各种药物。
表格2显示CB/ CP各种药物的比例。在所有药物中B/ CP比值在0.68 ~ 1.1之间,在0.5 ~ 10 μg/ml浓度范围内保持不变。KB用eq计算各种药物的值。8,列于表中3..平均KB,应用分别于静脉给药后15 min、1、4、8、24 h给予ITZ。的KB,应用先前获得的稳态值用于KTZ、CIM和NIZ (Takedomi等人,1998年;Yamano等人,1999年).因为KB,应用初步实验中,ENX、CPFX、CAM、VER和DLZ在静脉给药3 h后达到伪稳态(β相)KB,应用这些药物在静脉给药后3小时的值。
表格3.显示了fP各种各样的药物。fP从ITZ的0.0034到NIZ的0.96不等。表观的最大值K男朋友(KB,应用/ fP), ITZ最小值为6100,NIZ最小值为3.0。
因为KB,真正的VER和DLZ的值可能比KB,应用由于其肝提取比大,肝提取比确定计算KB,真正的值。真正的K男朋友VER和DLZ分别为600和73,是表观的4.9倍和4.6倍K男朋友值,分别。真正的K男朋友ENX、CPFX和CAM的值分别为9.3、11和36,非常接近表观K男朋友值。
分离的大鼠肝细胞摄取各种药物。
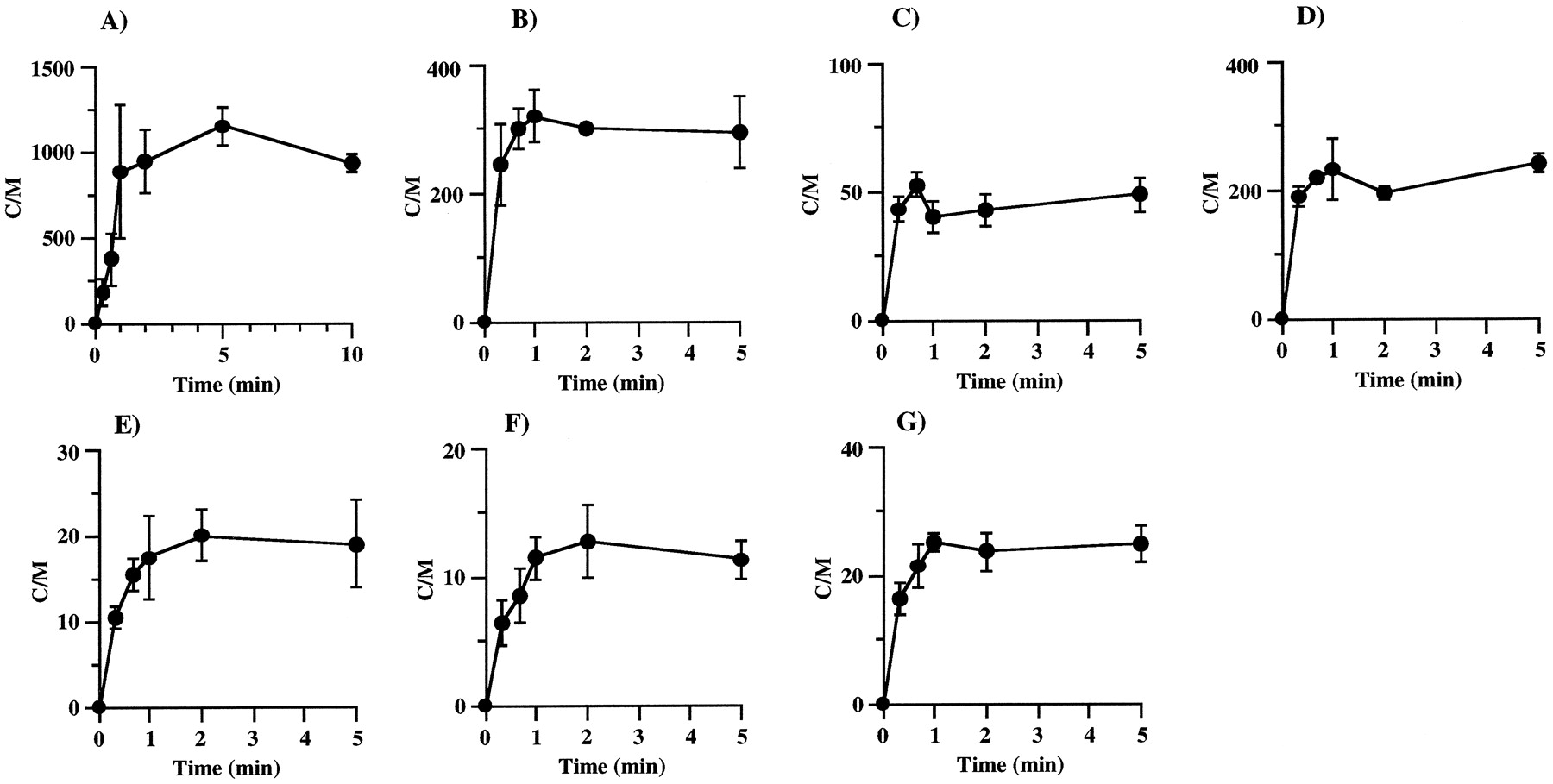
数字2显示了各种药物进入离体大鼠肝细胞的时间过程。KTZ、VER、DLZ、ENX、CPFX和CAM的吸收在5 min内达到平衡,而ITZ的吸收需要10 min。在浓度依赖性研究中,每种药物的孵育时间设定为达到平衡。

离体大鼠肝细胞摄取ITZ (A)、KTZ (B)、DLZ (C)、VER (D)、ENX (E)、CPFX (F)、CAM (G)的时间历程。
孵育液中药物浓度为1 μg/ml。每一个点和竖线代表平均值±标准差(n= 3)。
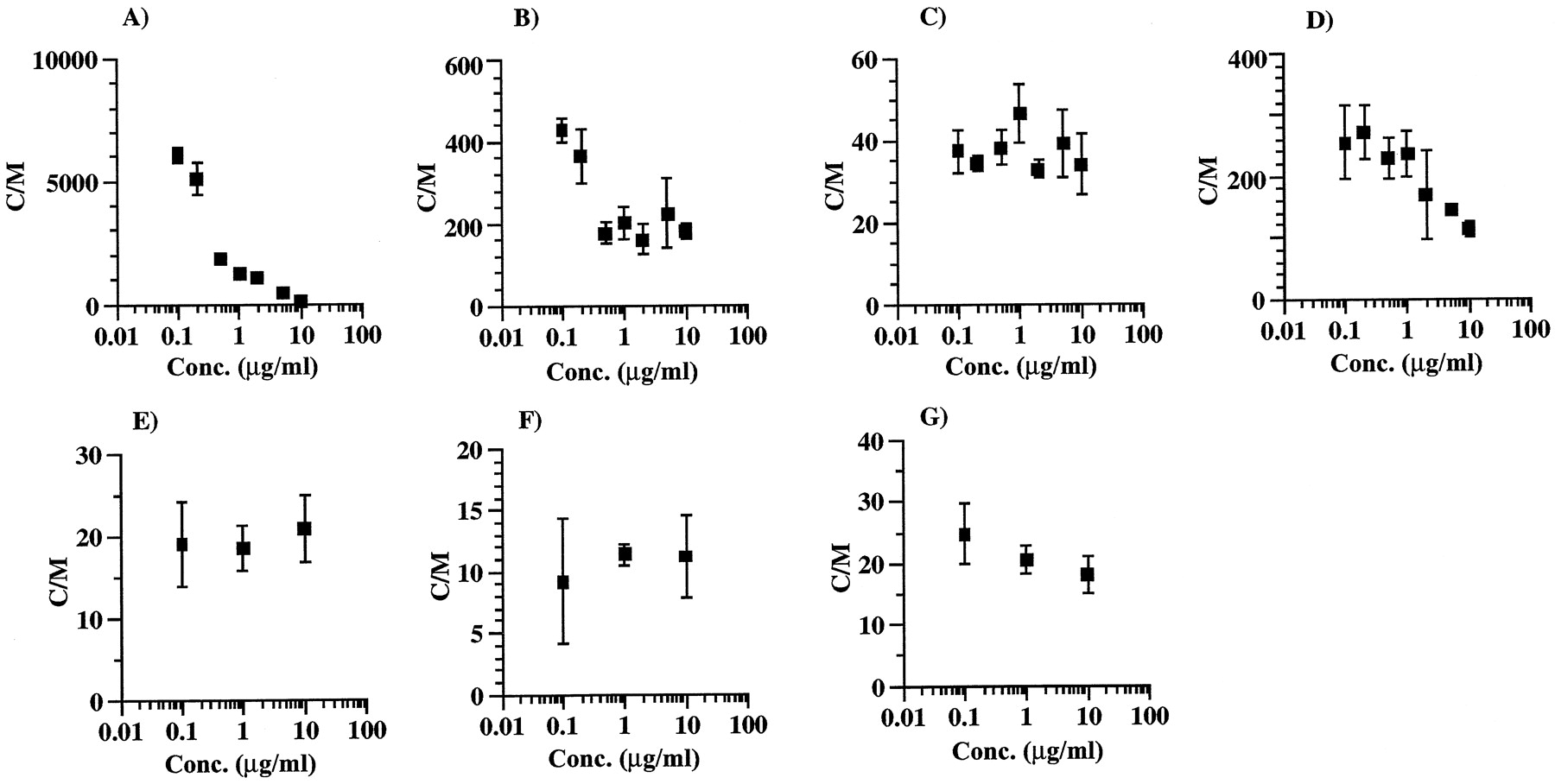
数字3.显示了不同药物进入离体大鼠肝细胞的浓度依赖性。ITZ的吸收表现出明显的浓度依赖性。在10 μg/ml和0.1 μg/ml浓度下,ITZ的C/M比分别为~ 200和6000。KTZ和VER的吸收浓度依赖程度小于ITZ, DLZ、ENX、CPFX和CAM的吸收浓度依赖程度可忽略不计。

分离的大鼠肝细胞摄取ITZ (A)、KTZ (B)、DLZ (C)、VER (D)、ENX (E)、CPFX (F)和CAM (G)的浓度依赖性。
ITZ和其他药物的孵育时间分别为10和5 min。每一个点和竖线代表平均值±标准差(n= 3)。
离体大鼠肝细胞在肝内分布与体外摄取的相关性。
药物的C/M比与大鼠离体肝细胞摄取的相关性K男朋友研究了大鼠肝脏中的值,以评估体外实验预测给药后肝脏中的浓度的有效性。在浓度几乎等于血浆中未结合浓度时的C/M比被用来研究C/M比与心肌梗死之间的相关性K男朋友除ITZ外,其他药物的值。由于给药速率为5.7和11.4 mg/h/体后,CIM和NIZ的解结合浓度分别为0.5 ~ 5和1 ~ 5 μg/ml,故采用添加浓度为3 μM (0.76 μg/ml)和3 μM (0.99 μg/ml)时CIM和NIZ (Nakamura等,1994),分别。大鼠静脉给药20 mg/kg后,血浆中ITZ的游离浓度在5 ~ 20 ng/ml范围内,但在如此低的浓度下,很难确定ITZ的C/M比。在本研究中,使用添加浓度为100 ng/ml时的C/M比,因为添加浓度为0.1和0.2 μg/ml时的C/M比没有显著差异,表明在较低浓度时C/M比恒定。数字4显示了C/M比率与K男朋友九种药物的价值。C/M比值的对数与K男朋友值(r= 0.981)。

离体大鼠肝细胞摄取C/M比与肝/血浓度比的相关性。
讨论
为了开发一种定量预测药物-药物相互作用风险的方法,有必要解决四个问题:1)预测抑制剂在肝脏中的分布非常重要,因为许多药物是通过载体介导的肝脏摄取系统(Meijer等人,1990年;山崎等人,1996)和肝脏中的游离浓度高于血浆中的游离浓度;2)由于临床上大部分药物为口服,门静脉内药物浓度高于体循环内药物浓度,因此有必要预测门静脉或肝静脉内抑制剂的浓度(Hoffman等人,1995;Tabata等人,1995;Fujieda等,1996);3)由于CYP3A4在肠道中的活性是在肝脏中的活性的一半,因此有必要预测药物-药物相互作用在肠道(空肠、回肠)和肝脏中的代谢过程。据报道,MDZ和环孢素在肠道中被CYP3A4代谢(Paine等人,1996年;Thummel等人,1996年);4) p -糖蛋白存在于肠道中,参与药物的分泌,因此有必要预测药物-药物相互作用对药物吸收过程的影响。
然而,我们认为,同时解决上述所有问题是困难的。为了定量评估与肝脏代谢抑制相关的药物-药物相互作用的程度,我们试图通过使用MDZ同时给药ITZ、KTZ、CIM和NIZ作为抑制剂来预测大鼠血浆中MDZ的浓度增加率(R) (Takedomi等人,1998年;Yamano等人,1999年).假设药物代谢的相互作用为竞争抑制型,血药浓度的增加比例可由下式估计: 方程10我和K我分别为抑制剂浓度和抑制常数。预测的与血浆中游离浓度的增长比率被低估,而与肝脏中游离浓度的增长比率与观测值非常接近。抑制剂的肝脏游离浓度与血浆游离浓度之比为>1,表明这些药物被集中摄取进入肝脏。由于代谢酶定位于肝细胞内的内质网,并被质膜与血液物理分离,因此肝脏内的游离浓度可能更适合定量预测血浆浓度的增长速度。在人体中,需要肝脏游离浓度的抑制剂。然而,很难直接测量肝脏游离浓度的抑制剂;因此,有必要采用一种方法来预测给药后肝脏中的浓度。我们试图预测给大鼠给药后肝脏中的浓度。首先,我们调查了K男朋友大鼠肝细胞摄取的C/M比。其次,我们研究了用C/M比值和血浆或血液中的浓度来预测肝脏中的浓度的可能性。
方程10我和K我分别为抑制剂浓度和抑制常数。预测的与血浆中游离浓度的增长比率被低估,而与肝脏中游离浓度的增长比率与观测值非常接近。抑制剂的肝脏游离浓度与血浆游离浓度之比为>1,表明这些药物被集中摄取进入肝脏。由于代谢酶定位于肝细胞内的内质网,并被质膜与血液物理分离,因此肝脏内的游离浓度可能更适合定量预测血浆浓度的增长速度。在人体中,需要肝脏游离浓度的抑制剂。然而,很难直接测量肝脏游离浓度的抑制剂;因此,有必要采用一种方法来预测给药后肝脏中的浓度。我们试图预测给大鼠给药后肝脏中的浓度。首先,我们调查了K男朋友大鼠肝细胞摄取的C/M比。其次,我们研究了用C/M比值和血浆或血液中的浓度来预测肝脏中的浓度的可能性。
由于离体大鼠肝细胞摄取的C/M比为药物摄取达到平衡时的值,因此,该模型具有较好的应用价值K男朋友当肝脏浓度与血浆或血液中的浓度平行时,在大鼠中的值。对于VER和DLZ,肝脏是主要消失器官,总清除以肝脏清除为主。静脉给药后VER和DLZ的总清除率分别为43.4±4.2和63.9±6.3 ml/min/kg(平均±sd)。n= 4),与肝脏血流速率相似。的KB,真正的VER和DLZ的值可能比KB,应用值,因为它们的肝脏提取率大。因此,我们计算了他们的肝提取率的AUC后,门户内和静脉给药,然后KB,真正的根据eq计算数值。7.静脉注射ENX和CPFX后的总清除率分别为38.7±2.0和51.9±14.4 ml/min/kg(平均±sd)。n= 4),但清除途径均为肝代谢和肾排泄,ENX和CPFX的肝脏清除率分别为12和25 ml/min/kg (戴维斯等人,1995年),远低于肝脏血流速率。因此,K男朋友肝清除率和肝血流率计算值与真实值接近K男朋友值。
在离体大鼠肝细胞中观察到ITZ、KTZ和VER的浓度依赖性摄取,并提示可饱和载体介导的摄取,而其他药物在离体大鼠肝细胞中的摄取不依赖于药物浓度。数字4表明药物的C/M比与真实值有良好的相关性K男朋友老鼠的价值观。因此,肝脏中的药物浓度(CH),可从大鼠离体肝细胞摄取的C/M和血液中的药物浓度预测,如eq。11:
脚注
将转载要求发送至:Katsuhiro Yamano,生物制药和药代动力学研究实验室,藤泽制药株式会社,1-6,鹿岛2栋,yodogawa区,大阪532-8514
- 使用的缩写有::
- AUC
- 浓度-时间曲线下的面积
- MDZ
- 咪达唑仑
- ITZ
- 伊曲康唑
- KTZ
- 酮康唑
- CIM
- 西咪替丁
- 凸轮
- 克拉霉素
- NIZ
- nizatidine
- C / M
- 细胞内浓度与培养基浓度之比
- K 男朋友
- 血液中肝脏浓度与游离浓度的比值
- ENX
- enoxacin
- CPFX
- 环丙沙星
- 凸轮
- 克拉霉素
- 版本
- 维拉帕米
- DLZ
- 地尔硫卓
- K P
- 肝脏/血浆浓度比
- AUC4
- 静脉注射后血药浓度-时间曲线下面积
- AUC光伏
- 门内给药后血浆浓度-时间曲线下面积
- K B
- 真实肝脏/血液浓度比
- K B,应用
- 肝/血浓度比
- 问H
- 肝脏血流率
- VH
- 肝容积
- fb
- 血液中游离的部分
- fP
- 等离子体中未结合的部分
- CB
- 血液浓度
- CP
- 血浆浓度
- R
- 增加的比例
-
- 收到了1999年2月16日。
- 接受1999年6月15日。
- 美国药理学和实验疗法学会








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}