

失调间充质细胞的生存途径严重纤维化肺病:Nintedanib疗法的效果
 Rajesh k Kasam
Rajesh k Kasam Geereddy b Reddy
Geereddy b Reddy 阿尼尔•g . Jegga
阿尼尔•g . Jegga Satish k Madala
Satish k Madala- 1医学院的儿科,辛辛那提,哦,美国辛辛那提大学
- 2分工肺药,辛辛那提儿童医院医学中心,辛辛那提,哦,美国
- 3生物化学、营养学研究所、海得拉巴,印度
- 4生物医学信息学,辛辛那提儿童医院医学中心,辛辛那提,哦,美国
凋亡清除myofibroblasts受损会导致疤痕组织的不断扩大在持续伤害肺部。然而,分子和细胞机制多种间充质细胞的凋亡间隙包括纤维细胞、成纤维细胞和myofibroblasts严重纤维化肺疾病如特发性肺纤维化(IPF)在很大程度上仍未知。我们分析了IPF的间充质细胞凋亡通路激活在TGFα-induced肺纤维化小鼠模型。我们发现,纤维细胞和肺纤维化病变myofibroblasts获得抗病性Fas-induced凋亡,和一个fda anti-fibrotic代理,nintedanib,有效地诱导凋亡的细胞死亡。支持,比较基因表达分析表明,apoptosis-linked基因网络同样IPF和特异表达TGFα-induced肺纤维化小鼠模型。TGFα小鼠接受nintedanib显示增加活跃的半胱天冬酶3-positive细胞纤维化病变,减少fibroproliferation和胶原蛋白的生产。此外,长期nintedanib疗法减毒纤维细胞积累,胶原沉积,在TGFα-induced肺纤维化肺功能下降。这些结果强调生存抑制通路的重要性和其他pro-fibrotic过程中各种类型的间充质细胞,表明TGFα小鼠模型相关测试anti-fibrotic药物单独或结合nintedanib。
介绍
特发性肺纤维化(IPF)是一种致命的肺纤维化疾病与异常激活成纤维细胞,导致其过度增殖、生存、积累、细胞外基质(ECM)和生产(艰苦的et al ., 2009)。仅在美国,这种疾病会影响大约200000人每年∼40000死。平均存活率是诊断后2 - 3年。因此,这种疾病每年造成更多的生命比许多类型的癌症(格里宾et al ., 2006;哈钦森et al ., 2014)。
两个美国食品和药物管理局(FDA)批准的药物,Ofev (nintedanib)和Esbriet (pirfenidone),可作为IPF患者的新疗法。尽管他们改善肺功能,其影响机制在很大程度上仍未知(王et al ., 2014;Richeldi et al ., 2014;内森et al ., 2017)。活跃的纤维发生期间,成纤维细胞增殖和transdifferentiate效应myofibroblasts,分泌ECM组件的远端肺的实质区域,导致肺泡破坏和受损的气体交换(黄和霍洛维茨,2014年;弗里曼et al ., 2017)。在一个典型的愈合反应,myofibroblasts积累损伤后迅速恢复屏障功能和启动的肺损伤修复反应。后,细胞凋亡清除阻止疤痕组织的形成(Desmouliere et al ., 1995)。然而,持续的损伤和过度生长因子的生产、抗凋亡间隙myofibroblasts和肺纤维化病变发展不断扩大(永利,2007;Marudamuthu et al ., 2015)。理解的分子控制dysfuction将为开发有效antifibrotic药物铺平了道路。
许多类型的间充质细胞,包括lung-resident成纤维细胞和纤维细胞、填充纤维损伤,提示他们转换为myofibrobalsts和永久的疤痕组织的形成。Myofibroblasts终末分化,纺锤状细胞表达α-smooth肌肉肌动蛋白(αSMA)和积累在纤维化病变达到抗凋亡表型和无情地分泌ECM组件(Desmouliere et al ., 1995)。最近的发现表明生存通路的激活在myofibroblasts肺纤维化的发病机制。特别是myofibroblasts隔绝IPF患者的肺部表现出抗Fas-mediated细胞凋亡(田中et al ., 2002)。细胞凋亡监管机构,如细胞凋亡抑制蛋白(XIAP),报价,Blc-2,和细胞FLICE抑制蛋白(c-FLIP),特异表达(Golan-Gerstl et al ., 2012;Ajayi et al ., 2013;Safaeian et al ., 2014),和一些生长因子和转导通路已经被证明来激活各种prosurvival myofibroblasts通路(Thannickal和霍洛维茨,2006年;霍洛维茨et al ., 2012;Nho et al ., 2013)。然而,精确的基因网络启动和维护在间充质细胞抗凋亡仍然很大程度上定义。理解这些生存通路间充质细胞积聚在纤维化病变会加速新药的发现在IPF (格拉瑟et al ., 2016)。
纤维细胞是骨骨髓来源间充质细胞表达造血细胞表面标记CD45和间叶细胞特异性标记,如胶原蛋白和波形蛋白(抗起球et al ., 2009;大师et al ., 2013;Sontake et al ., 2019)。在损伤和纤维化,纤维细胞积聚在不同的肺的病理区域(Madala et al ., 2014 b)。评价IPF活检和肺纤维化小鼠模型表明他们与myofibroblasts积累成熟的纤维化病变(摩尔et al ., 2005,2006年;抗起球et al ., 2007;Moeller et al ., 2009;抗起球歌篾,2014),他们的过度积累增加fibroproliferation和ECM沉积(Kleaveland et al ., 2014;Sontake et al ., 2015)。纤维细胞和myofibroblasts坚持在一起,相互保持成熟的纤维化病变,但我们不知道如何生存通路损害他们的间隙。阐明这一过程是至关重要的为开发治疗策略,目标多种间充质细胞和诱导纤维化病变的回归。
Nintedanib (BIBF 1120)是一个indolinone导数与ATP结合口袋的血管内皮生长因子受体(VEGFR),血小板源生长因子受体(PDGFR)和成纤维细胞生长因子受体(FGFR),阻止他们的酪氨酸激酶介导的活动(Hilberg et al ., 2008)。Nintedanib已经证明有效的减少纤维化的负担和IPF患者肺功能下降Richeldi et al ., 2014)。不幸的是,行动的机制仍然是难以捉摸的。它已被证明会减弱胶原蛋白沉积在bleomycin-induced肺纤维化(Wollin et al ., 2014;李et al ., 2017)和胶原蛋白水平降低的小鼠模型类风湿性arthritis-associated间质性肺疾病(Redente et al ., 2018)。它可以减弱纤维母细胞增殖,迁移,fibroblast-to-myofibroblast转换和ECM的合成在体外(Wollin et al ., 2015;黄et al ., 2016)。同时,nintedanib显示诱导成纤维细胞的自噬途径从肺部孤立IPF患者(Rangarajan周二et al ., 2016)。虽然纤维细胞和lung-resident myofibroblasts已被证明积聚在纤维化病变,导致积累的机制仍然未知。理解的分子行动nintedanib作为anti-fibrotic治疗是至关重要的开发更有效的治疗,单独或结合nintedanib采取行动提高IPF患者的存活率。
材料和方法
TGFα-Induced肺纤维化小鼠模型和Nintedanib疗法
代TGFα-overexpressing老鼠前面描述的(艰苦的et al ., 2004)。克拉拉特异性protein-rtTA+ / -(CCSP-rtTA)老鼠穿过杂合的(TetO)7小鼠巨细胞病毒TGFα生产bitransgenic CCSP / TGFα老鼠。诱导TGFα表达、转基因老鼠与强力霉素(阿霉素)包含周(62.5毫克/公斤)(Madala et al ., 2014 c)。男性和女性在10到16周的年龄性别小鼠被用于所有的研究。他们被安置在特定的无菌条件下,依照协议机构批准的动物保健和使用委员会辛辛那提儿童医院研究基金会的。Nintedanib(开曼化学,安阿伯市,美国)准备在每天新鲜的车辆(0.5%羧甲基纤维素)治疗。纤维化是由overexpressing TGFα3周,和在过去的5天,车辆或nintedanib(60毫克/公斤,一天一次)是由口腔填喂法描述(Madala et al ., 2016 b)。慢性干预研究,所有组的老鼠开始对阿霉素总7周。本周初4广泛纤维化时,控制和TGFα老鼠处理车辆或nintedanib最后4周(Sontake et al ., 2017)。Non-TGFα表达小鼠与车辆使用阿霉素治疗作为对照组,以确定车辆的肝纤维化程度和药物治疗组。
人类和小鼠肺主间充质细胞培养
人类和小鼠肺间质细胞培养所描述的准备(Sontake et al ., 2017,2018年)。孤立纤维细胞和lung-resident myofibroblasts、肺间质细胞收获和孵化anti-CD45微冰的15分钟(Miltenyi研究,赤褐色,CA,美国)。与无菌缓冲洗两次后,细胞被加载到磁列(Miltenyi研究)和筛选了适量无菌缓冲区的存在和缺乏磁场分离的细胞(CD45负细胞;lung-resident myofibroblasts或者绑定到列(CD45+ ve细胞;纤维细胞)。间充质细胞纯度子集确定使用流式细胞仪(≥96%)Madala et al ., 2014 b)。人类和小鼠间充质细胞在DMEM培养与10%的边后卫和IMDM 5%的边后卫媒体,分别。通道1 - 5之间的主要细胞用于实验。
RNA提取和实时PCR
从孤立的细胞和肺组织总RNA制备使用RNeasy迷你包(试剂盒科学、瓦伦西亚、钙、美国)作为描述(Madala et al ., 2012)。互补DNA制备、实时PCR进行使用CFX384触摸实时PCR检测系统和SYBR绿色超级混合(Bio-Rad、大力神、钙、美国)。目标基因记录在每个样本被归一化鼠标次黄嘌呤鸟嘌呤phosphoribosyl转移酶(产生Hprt)或人类beta-actin。表1,2列出了实时引物用于这项研究。
表1

表1。列表中鼠标rt - pcr引物用于这项研究。
表2

表2。人类的rt - pcr引物用于这项研究。
免疫印迹
小鼠肺组织或主lung-resident myofibroblasts和纤维细胞治疗DMSO溶液或nintedanib细胞溶解使用里帕裂解缓冲补充蛋白酶和磷酸酶抑制剂(美国丹佛市细胞信号技术)。总蛋白量化使用BCA试剂盒(热费希尔科学、沃尔瑟姆,MA)和等量的蛋白质可溶性部分受到了sds - page凝胶所描述的4 - 12% (辛格et al ., 2017)。主要的抗体使用贝克(# 12105)和伯灵顿(# 2772)从细胞信号技术(丹佛、有限公司、美国),和Collagen1α(美国圣克鲁斯生物技术、达拉斯、TX)和Gapdh (Bethyl实验室)。量化使用卷集成函数执行的磷光成像软件,Multigage(富士胶片,瓦尔哈拉殿堂,纽约,美国)作为描述(Madala et al ., 2016 a)。
IncuCyte变焦半胱天冬酶3/7凋亡测定
动能半胱天冬酶估计3/7的活动是使用实时成像系统执行IncuCyte变焦(埃森生物科学,安阿伯市,美国)。激活细胞发生凋亡的caspase-3/7死亡劈开caspase-3/7衬底产生核绿色荧光(caspase-3/7绿色细胞凋亡测定试剂[埃森生物科学])。主要lung-resident myofibroblasts或纤维细胞从正常或纤维化肺组织准备和培养12-well confluency板到50 - 60%。在一夜之间日益增长的低血清MEM媒体,他们已经适应了low-serum条件。然后他们被媒体包含半胱天冬酶处理3/7绿色细胞凋亡测定试剂在最后5μM /毫升的浓度或半胱天冬酶3/7绿色细胞凋亡测定试剂和anti-Fas抗体(BD生物科学)的最终浓度250 ng / mL。延时荧光成像技术进行使用IncuCyte变焦系统(埃森生物科学);每在20×9图像放大收集每2 h为24 - 48 h。绿色的平均数对象产生的细胞凋亡测定使用IncuCyte放大软件2015 a。
免疫组织化学和细胞计数
Formalin-fixed肺部分与抗体制备和应用主动caspase-3(# 9664, 1∶细胞信号技术)和ki - 67(1:6 00 # 12202年,细胞信号技术)作为描述(Sontake et al ., 2015)。从每个肺部分,5 - 10高倍率(40×)胸膜下的图像区域得到随机使用Nikon-Ni-E直立的显微镜。活跃caspase-3或ki - 67阳性细胞数使用MetaMorph成像软件(分子器件、桑尼维尔,美国)作为描述(Sontake et al ., 2015)。
组织学、胸膜厚度测量和肺功能测试
肺膨胀并使用10%缓冲福尔马林固定和染色与梅森的三色的前面描述的(Sontake et al ., 2017)。测量胸膜厚度、五个随机图像(40×)收集从每个老鼠使用亮视场显微镜(徕卡微系统)。胸膜厚度测量MetaMorph成像软件的使用距离测量功能(v6.2:分子器件、桑尼维尔,美国)如前所述(Sontake et al ., 2017)。肺功能测试是由一个电脑福莱希通风系统(SCIREQ,蒙特利尔,加拿大)如前所述(Sontake et al ., 2015)。
BrdU扩散分析
主要lung-resident纤维母细胞增殖是由BrdU细胞增殖试验装备(美国丹佛市细胞信号技术)作为描述(Sontake et al ., 2017,2018年)。简而言之,主要lung-resident成纤维细胞治疗用DMSO溶液或nintedanib(0.1、0.5和1μM) 24小时然后孵化与另一个24小时BrdU标记的解决方案以及nintedanib或DMSO溶液。细胞被固定后24 h BrdU标记,执行和immunodetection BrdU根据制造商的协议。改变在扩散计算折叠不同控制权通过测量吸光度在450海里。
计算分析
我们进行直接比较的差异表达基因(度)与细胞凋亡与IPF TGFα老鼠阿霉素为3周。我们使用以前公布的转录组数据集(GSE53845) (DePianto et al ., 2015)来源于肺活检的分析40 IPF患者和8个健康对照组和可用的国家生物技术信息中心(NCBI)基因表达综合(GEO) (巴雷特et al ., 2007)。这IPF基因签名与度从TGFα对阿霉素小鼠3周(Sontake et al ., 2015)。IPF之间的相交,和抑制基因和TGFα对阿霉素小鼠3周然后进行功能富集分析使用的ToppFun应用ToppGene套件(陈et al ., 2009)。对于网络的表示选择大大丰富生物过程(细胞凋亡在这种情况下),Cytoscape应用程序(香农et al ., 2003)使用。
流式细胞术
总肺细胞从肺间质文化控制的老鼠和老鼠TGFα处理车辆或nintedanib用于染色CD45如前所述(Madala et al ., 2014 b)。数据是使用BD FACSCANTO II (BD生物科学)和分析了使用FACSDIVA软件(BD生物科学)。
统计分析
所有的数据进行了分析使用棱镜(版本7.02;美国GraphPad拉霍亚,CA)。学生的t以及被用来比较两个实验小组。单向方差分析与Sidak多重比较被用来比较不同实验小组,和双向方差分析比较组之间的独立变量。数据被认为是显著的p值小于0.05。
结果
Nintedanib诱发凋亡清除Lung-Resident Myofibroblasts
在IPF ECM-producing myofibroblasts积聚在肺纤维化病变发展抗细胞凋亡(Frankel et al ., 2006;Thannickal和霍洛维茨,2006年)。Nintedanib被证明减弱纤维母细胞增殖、迁移和转换,但其影响凋亡间隙没有探索(Wollin et al ., 2015)。评估对凋亡nintedanib间隙的影响,lung-resident myofibroblasts IPF患者培养的半胱天冬酶的存在3/7衬底共轭绿色荧光团和处理车辆或nintedanib(0.5和1μM)。我们观察到显著增加裂解caspase-3活动,因此,更多的凋亡细胞(绿色)在lung-resident myofibroblasts对待nintedanib相比单独车辆(图1 a, B)。
图1
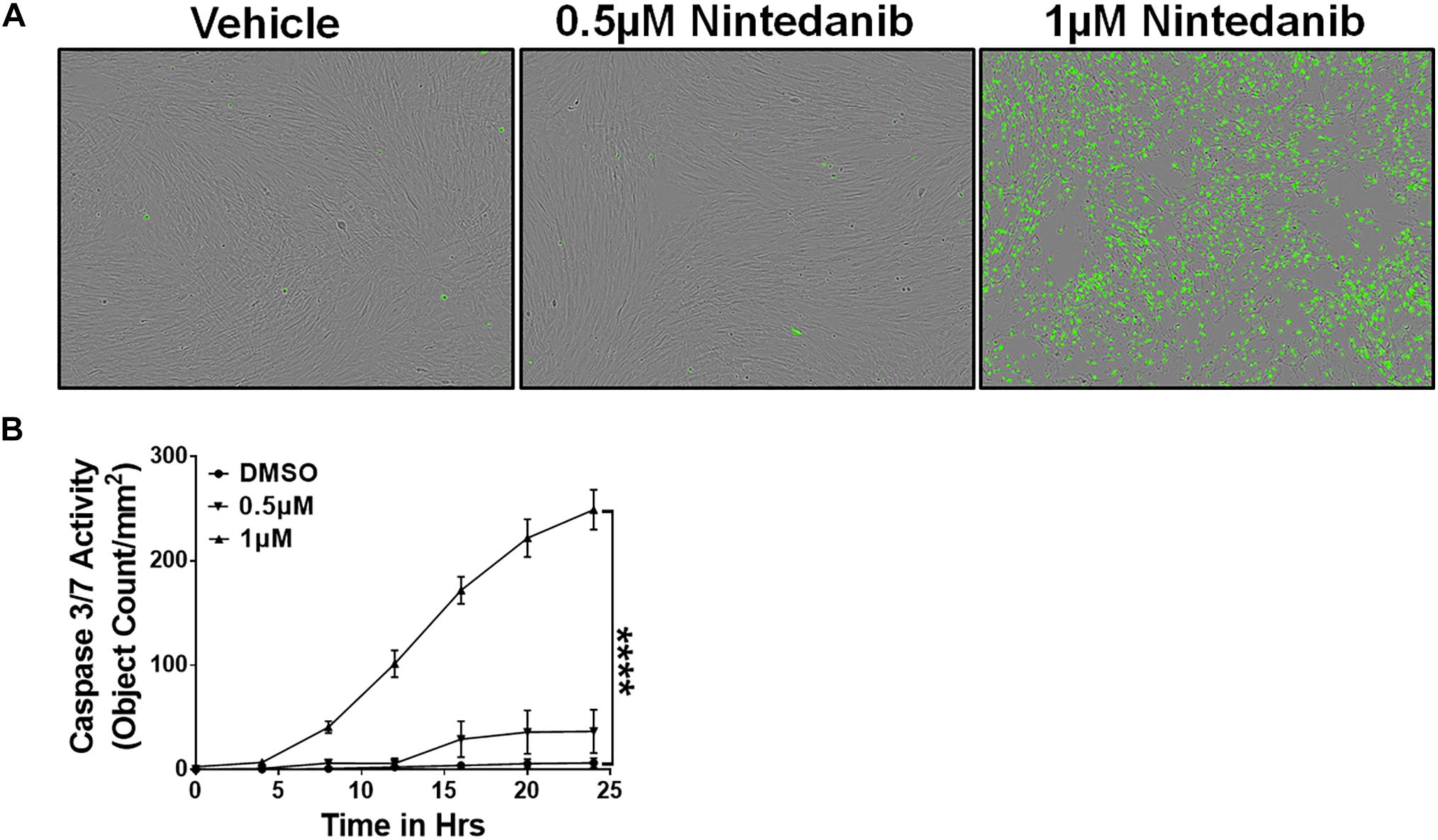
图1所示。Nintedanib治疗触发凋亡清除lung-resident myofibroblasts IPF。主要lung-resident myofibroblasts (CD45- - - - - -Col1+)与IPF隔离肺纤维母细胞文化通过与anti-CD45负选择磁珠。(一)代表图像的凋亡细胞(绿色,活跃caspase-3/7-positive) lung-resident myofibroblasts处理车辆或nintedanib(0.5和1μM) 24 h。(B)量化的凋亡细胞(绿色,活跃caspase-3/7-positive) lung-resident myofibroblasts处理车辆或nintedanib(0.5和1μM) (n= 4)。双向方差分析与Sidak多个测试是用来测量比较显著的差异。提出了数据均值±SEM (n= 4)。* * * *P< 0.00005。
此外,我们孤立lung-resident myofibroblasts TGFα和控制老鼠的肺文化阿霉素与媒体或4周和治疗anti-Fas抗体。像IPF myofibroblasts,抗细胞凋亡明显大于lung-resident myofibroblasts TGFα小鼠比正常小鼠(图2 a, B)。正如所料,anti-Fas抗体治疗导致了更多的细胞凋亡lung-resident myofibroblasts孤立从正常肺。值得注意的是,抗凋亡间隙坚持lung-resident myofibroblasts隔绝TGFα老鼠甚至在anti-Fas抗体的存在(图2 a, C)。确定nintedanib对细胞凋亡的影响,lung-resident myofibroblasts孤立TGFα小鼠肺纤维化的治疗与车辆或nintedanib和裂解caspase-3活动测量。结果,所示图2 d证明nintedanib治疗变弱凋亡抵抗lung-resident myofibroblasts TGFα小鼠对阿霉素4周。
图2
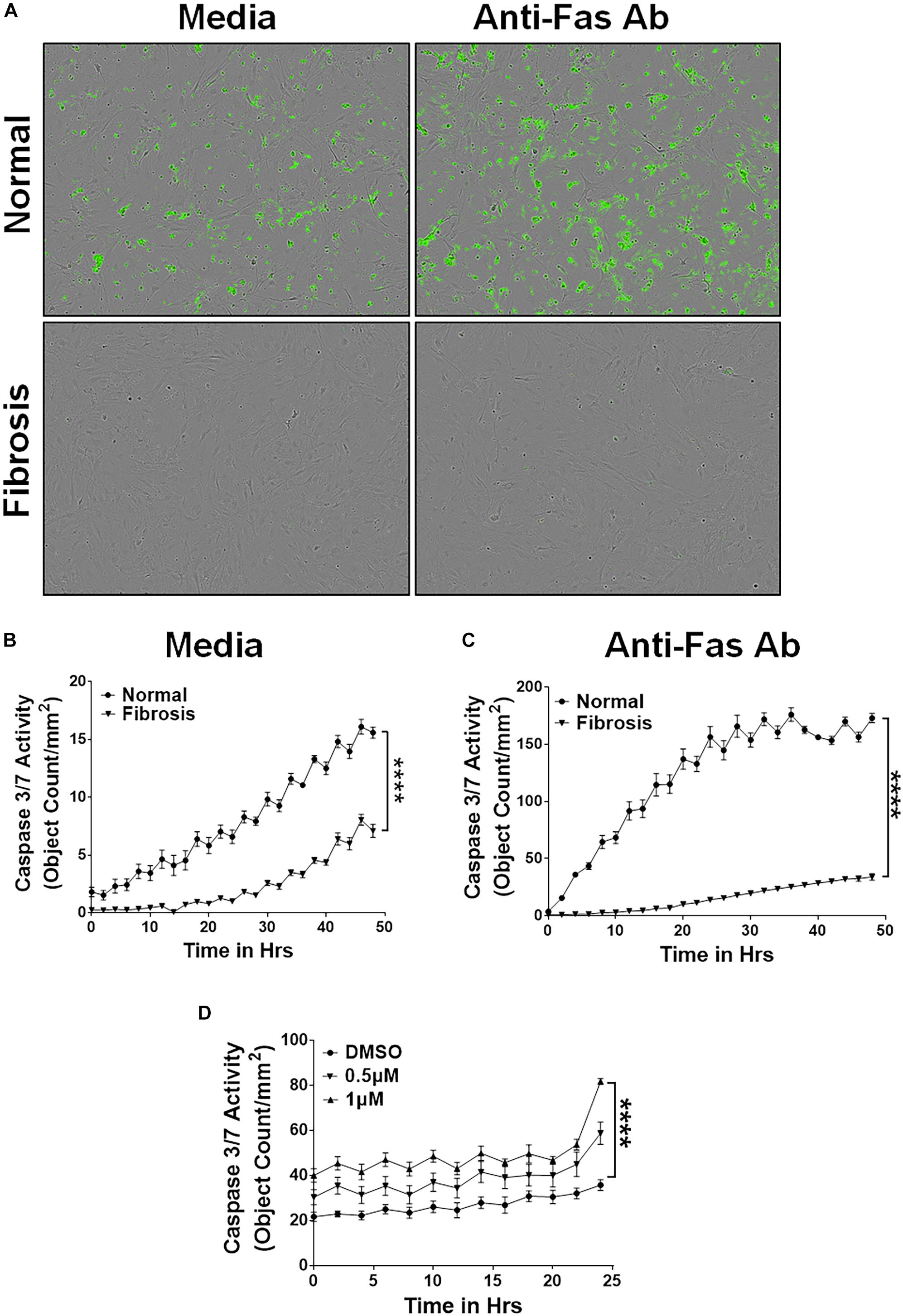
图2。期间积累的Nintedanib触发间隙apoptotic-resistant lung-resident myofibroblasts TGFα-induced肺纤维化。主要lung-resident myofibroblasts (CD45- - - - - -Col1+)与肺纤维母细胞正常小鼠的文化和TGFα转基因老鼠在阿霉素4周的负选择anti-CD45磁珠。(一)代表图像的凋亡细胞(绿色,Caspase-3/7-positive) lung-resident myofibroblasts培养与媒体或FasL (250 ng / ml) 24 h。(B)量化的凋亡细胞(绿色,Caspase-3/7-positive) lung-resident myofibroblasts孤立从正常和TGFα小鼠肺纤维化的阿霉素与媒体4周和治疗。(C)量化的凋亡细胞(绿色,Caspase-3/7-positive) lung-resident myofibroblasts孤立从正常和TGFα小鼠肺纤维化的阿霉素与FasL 4周和治疗(250 ng / ml) 48 h。(D)量化的凋亡细胞(绿色,Caspase-3/7-positive) lung-resident myofibroblasts孤立TGFα小鼠肺纤维化的对阿霉素4周和治疗与车辆或nintedanib(0.5和1μM) 24 h。双向方差分析与Sidak多个测试是用来测量比较显著的差异。所有数据提出了均值±SEM (n= 4)。* * * *P< 0.00005。
Nintedanib减弱纤维细胞生存表型
他人,我们已经表明,纤维细胞积聚在肺纤维化病变的IPF患者和TGFα转基因小鼠(Moeller et al ., 2009;Madala et al ., 2014 b)。我们假设增加纤维细胞生存会解释他们增加积累严重纤维化肺病。我们从肺孤立的纤维细胞的文化控制和TGFα转基因老鼠在阿霉素和培养4周半胱天冬酶的存在3/7量化半胱天冬酶底物3/7的活动。支持我们的假设,纤维细胞孤立TGFα转基因小鼠的肺纤维化病变比纤维细胞抗凋亡与正常肺(图3一)。测试Fas-mediated纤维细胞的凋亡,我们对待他们anti-Fas抗体和量化的凋亡细胞。我们发现降低易感性在纤维细胞与肺纤维化病变,而不是正常的(图3 b)。确定nintedanib纤维细胞发生凋亡,我们测量了裂解半胱天冬酶活动3/7纤维细胞纤维化肺的IPF患者和TGFα转基因小鼠。裂解半胱天冬酶3/7 nintedanib-treated纤维细胞的活动大大增强IPF (图3 c)和TGFα老鼠(图3 d)车辆相比,治疗。
图3
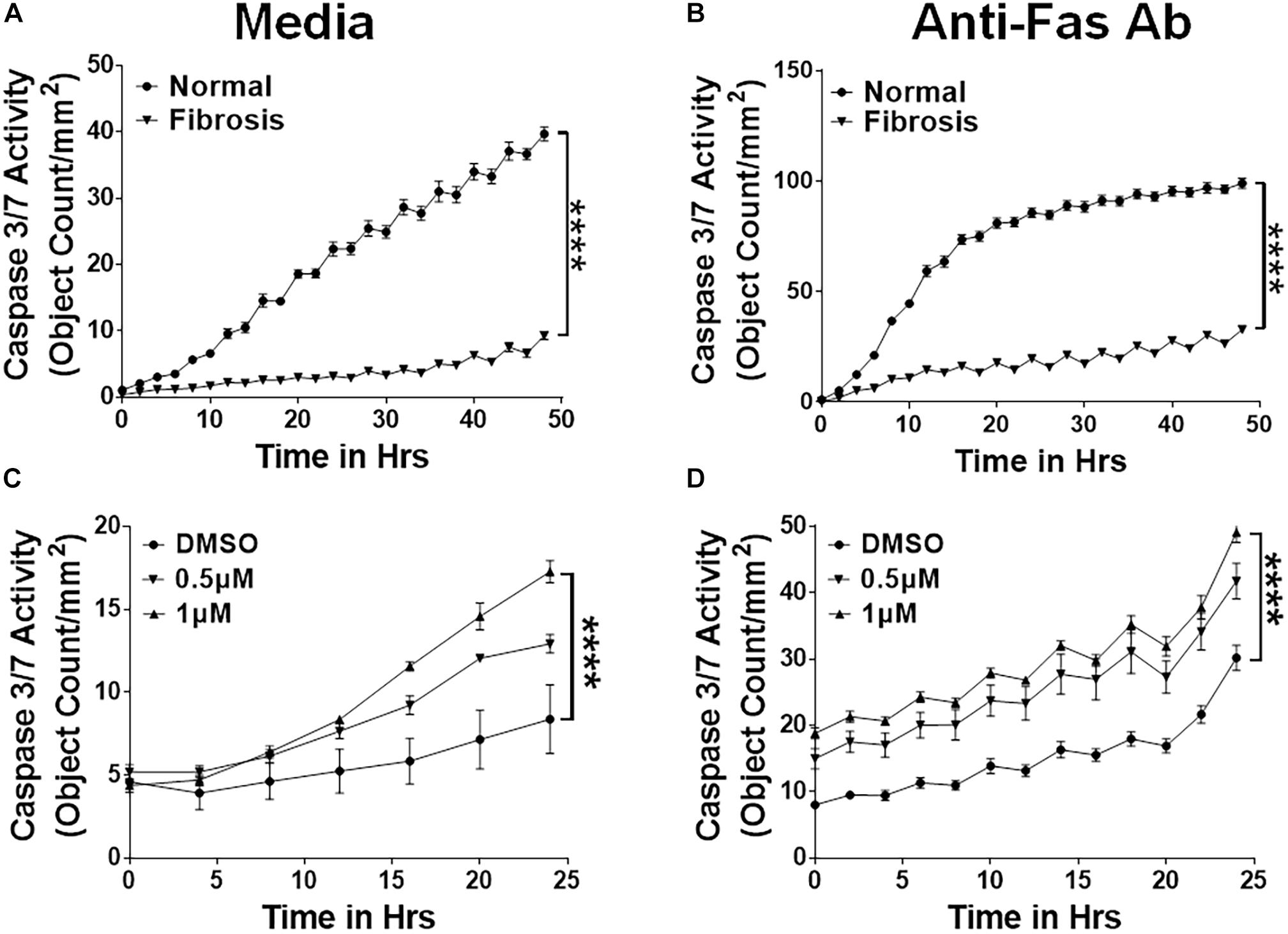
图3。期间积累的Nintedanib apoptotic-resistant纤维细胞的触发间隙TGFα-induced肺纤维化。主肺纤维细胞(CD45+Col1+)与肺间质文化的正常和TGFα转基因老鼠在阿霉素4周的积极的选择与anti-CD45磁珠。(一)凋亡细胞的量化(绿色,主动caspase-3/7-positive)从正常纤维细胞分离和TGFα小鼠肺纤维化的阿霉素与媒体(4周和治疗n= 4)。(B)凋亡细胞的量化(绿色,主动caspase-3/7-positive)从正常纤维细胞分离和TGFα小鼠肺纤维化的阿霉素与FasL 4周和治疗(250 ng / ml) 48 h (n= 4)。(C)量化的凋亡细胞(绿色,活跃caspase-3/7-positive)从IPF肺纤维细胞分离和处理车辆或nintedanib(0.5和1μM) (n= 4)。(D)凋亡细胞的量化(绿色,主动caspase-3/7-positive)在纤维细胞隔离TGFα小鼠肺纤维化的阿霉素与车辆或4周和治疗nintedanib(0.5和1μM) (n= 4)。双向方差分析与Sidak多个测试是用来测量比较显著的差异。所有数据都意味着±SEM。* * * *P< 0.00005。
IPF之间的重叠在Apoptosis-Linked基因的表达和TGFα模型
演示TGFα转基因小鼠模型的相关性评估的角色间充质细胞生存表型IPF发病机理,我们进行了一个比较apoptosis-linked基因的表达分析在IPF患者的肺部和TGFα老鼠。我们以前的研究表明,3周后在阿霉素,TGFα转基因老鼠发展纤维化病变,肺与成纤维细胞激活变化显著相关的记录(Sontake et al ., 2017)。这里,我们执行一个浓缩分析基因的转录,积极与数据集的IPF (DePianto et al ., 20153周)和TGFα老鼠阿霉素(Sontake et al ., 2015)。图4一显示重叠的表达提供成绩单的肺纤维化。特别是,维恩分析显示257年和183年基因转录,同样,或衰减与IPF TGFα老鼠阿霉素为3周。当我们执行一个使用这种重叠基因集富集分析,我们发现一个浓缩apoptosis-linked基因(图4 b)。
图4
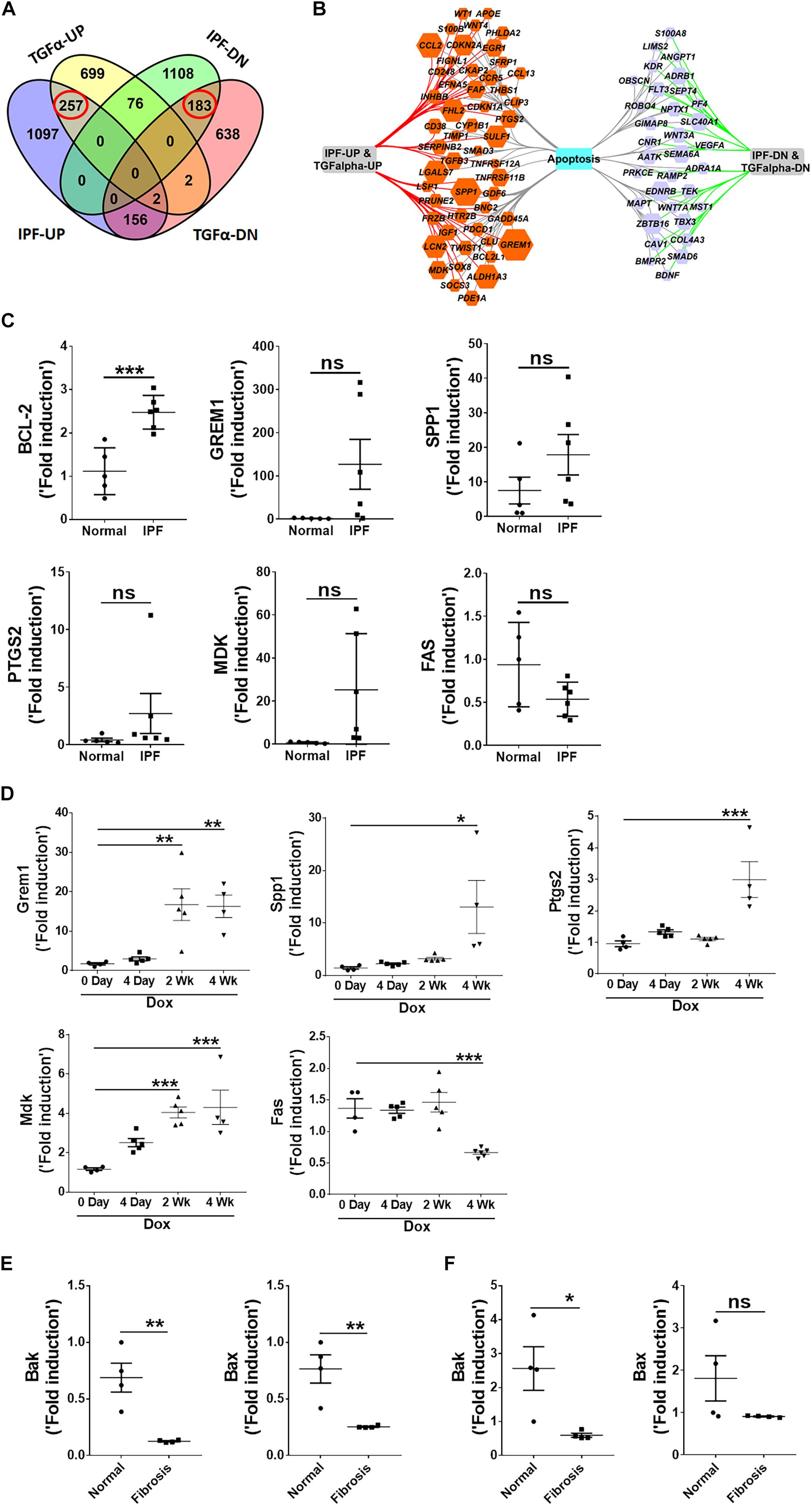
图4。Apoptosis-linked基因也同样在IPF特异表达和TGFα-induced肺纤维化小鼠模型。(一)维恩图显示了差异表达基因在IPF肺部和TGFα老鼠模型。虚线框表示基因调节基因(257)或表达下调(183)在IPF和TGFα老鼠阿霉素为3周。(B)丰富和apoptosis-linked基因同样在IPF特异表达和TGFα老鼠对阿霉素使用Cytoscape 3周了。橙色的调节基因是六边形,表达下调的基因在紫色的六边形。六角大小正比于叠化IPF的基因表达。(C)量化的bcl - 2, GREM1, SPP1、PTGS2 MDK, FAS rt - pcr基因转录的IPF肺(n= 5 - 6)。未配对学生的t以及用于测量组之间的统计学意义。(D)量化的Grem1、Spp1 Ptgs2 Mdk, rt - pcr和Fas成绩单的TGFα老鼠的肺在阿霉素0,4天,2周,4周(n= 4 - 6)。单向方差分析与Sidak多个比较测试被用来衡量一个显著差异。(E)量化的贝克和伯灵顿成绩单lung-resident myofibroblasts (CD45- - - - - -Col1+)从肺间质细胞培养分离从正常和TGFα老鼠阿霉素为4周(n= 4)。(F)量化的贝克和伯灵顿在肺纤维细胞(CD45成绩单+Col1+)从肺间质细胞培养分离从正常和TGFα老鼠阿霉素为4周(n= 4)。所有数据提出了均值±SEM。*P< 0.05,* *P< 0.005 * * *P< 0.0005;ns,不重要。
验证这一发现,我们使用rt - pcr分析apoptosis-linked基因RNA的表达与正常和IPF肺。我们观察到抗凋亡基因的表达增加,如Bcl2 Grem1, Spp1, Ptgs2,和Mdk pro-apoptotic基因的差别,对这些如FAS, IPF肺(图4 c)。我们也分析了apoptosis-linked基因的表达在肺部的TGFα转基因老鼠在强力霉素和观察到的进步增加Grem1成绩单,Spp1, Ptgs2,和在TGFα-induced Mdk肺纤维化,Fas表达显著下调TGFα老鼠阿霉素为4周(图4 d)。
确定pro-apoptotic基因表达间充质细胞中特异表达TGFα-induced肺纤维化,我们使用rt - pcr量化pro-apoptotic基因表达在lung-resident myofibroblasts和纤维细胞隔离肺的正常和TGFα老鼠对阿霉素4周。我们观察到pro-apoptotic基因的表达减少,如贝克和伯灵顿,在lung-resident myofibroblasts和纤维细胞孤立肺纤维化(图4 e, F)。
Nintedanib诱发Pro-apoptotic基因表达在纤维细胞和Lung-Resident Myofibroblasts
调查如何nintedanib凋亡,我们测量pro-apoptotic基因表达变化在lung-resident myofibroblasts与车辆或nintedanib纤维细胞治疗。Nintedanib治疗导致upregulation pro-apoptotic基因,如贝克和伯灵顿,lung-resident myofibroblasts (图5一个)和纤维细胞(图5 b)与肺纤维化病变TGFα小鼠对阿霉素4周。
图5
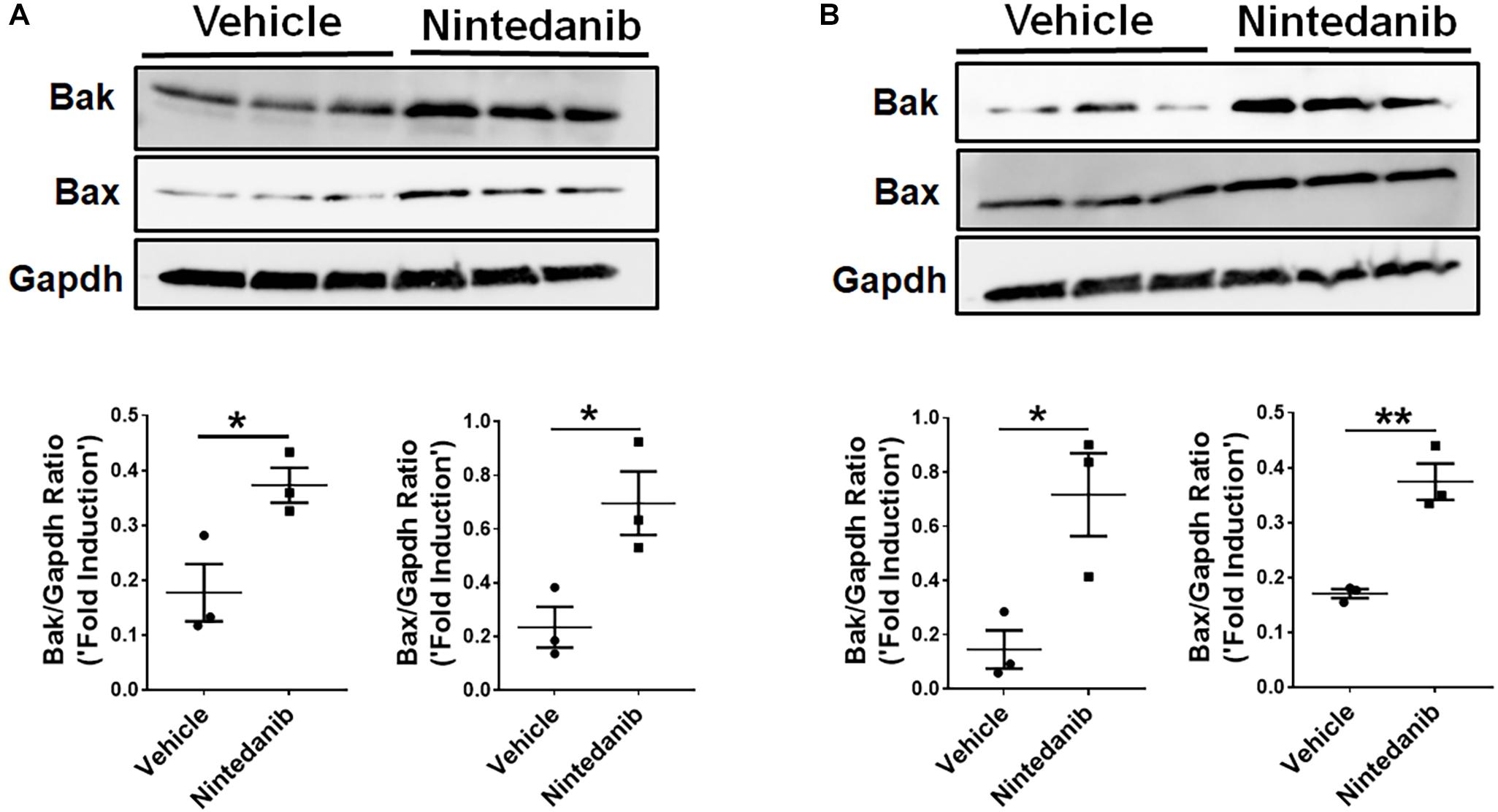
图5。Nintedanib诱发pro-apoptotic基因表达间充质细胞的TGFα纤维化的老鼠。主要lung-resident myofibroblasts (CD45- - - - - -Col1+)和纤维细胞(CD45+Col1+)隔绝TGFα小鼠的肺间质细胞培养4周的阿霉素-选择使用anti-CD45磁珠。(一)免疫印迹分析贝克,伯灵顿,Gapdh纤维化lung-resident myofibroblasts处理车辆或nintedanib(1μM)量化h。72年,贝克和伯灵顿规范化Gapdh加载控制。(B)免疫印迹分析贝克,伯灵顿,Gapdh纤维化肺纤维细胞处理车辆或nintedanib(1μM)量化h。72年,贝克和伯灵顿规范化Gapdh加载控制。未配对学生的t以及用于测量组之间的统计学意义。所有数据提出了均值±SEM (n= 3)。*P< 0.05,* *P< 0.005。
在活的有机体内Nintedanib治疗导致增加细胞凋亡和减少在间充质细胞增殖和ECM生产
进一步建立proapoptotic和anti-fibrotic nintedanib的影响在活的有机体内,TGFα转基因的老鼠喂Dox-containing 3周的食物最后5天治疗与车辆或nintedanib 60毫克/公斤(图6)。然后我们进行积极caspase-3疣状石蜡肺部分,发现显著增加活跃caspase-3-positive细胞从老鼠nintedanib-treated纤维化病变(图6 b, C),这也表达了显著减少ECM基因转录,如胶原蛋白1α和fibronectin1 (图6 d),比vehicle-treated TGFα老鼠。
图6
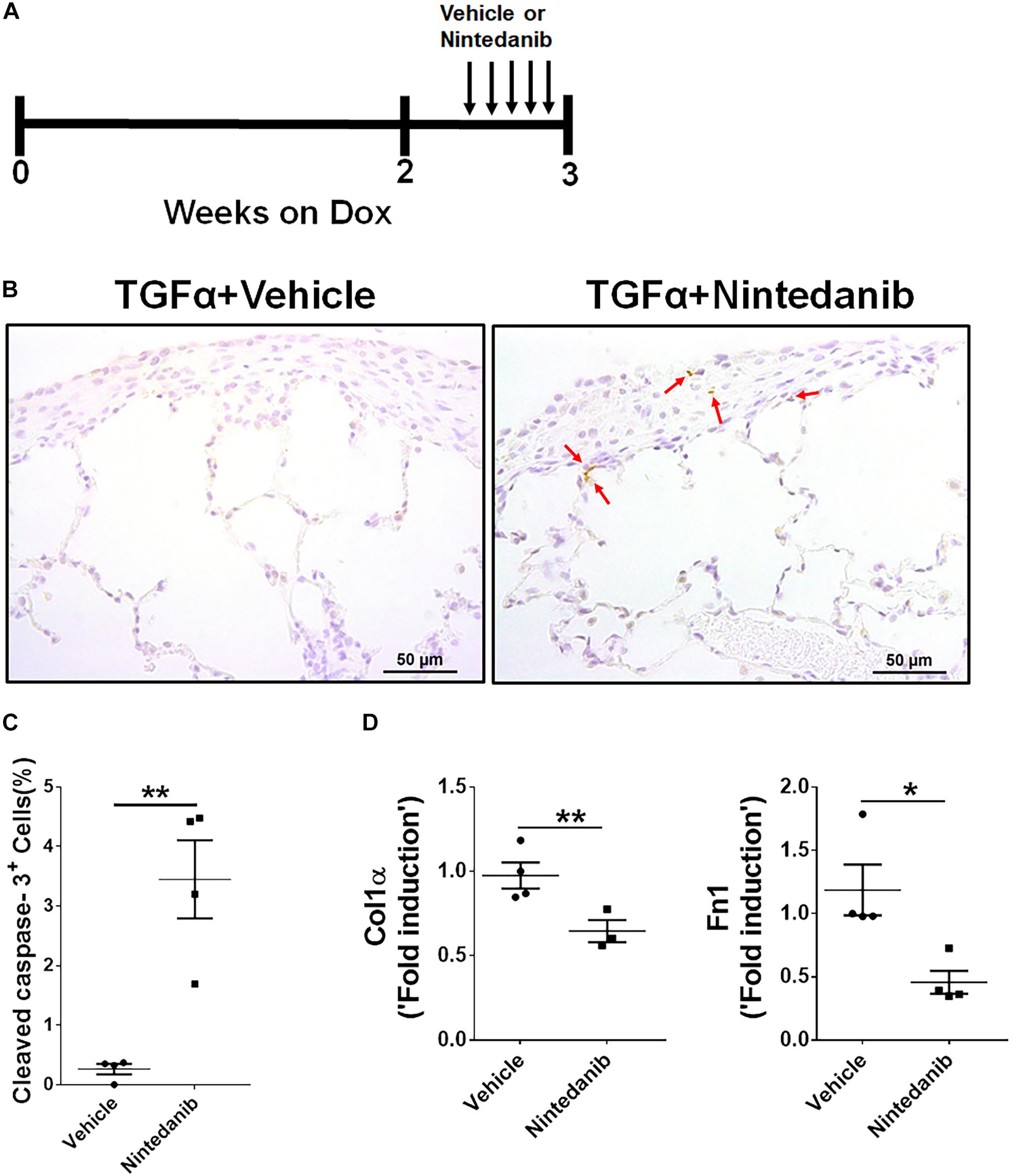
图6。Nintedanib凋亡,减少ECM生产期间TGFα-induced肺纤维化。(一)示意图说明在活的有机体内nintedanib治疗协议。TGFα老鼠强力霉素(阿霉素)3周治疗与车辆或nintedanib(60毫克/公斤)在过去的5天每天一次强力霉素。(B)从车辆或肺组织化学染色部分nintedanib-treated TGFαcaspase-3 Ab老鼠使用活跃。图像代表肺胸膜下区(n= 4 /组)。酒吧规模:50μm。(C)量化的活跃caspase-3百分比+细胞从车辆或胸膜下区nintedanib-treated组(n= 4 /组;5代表图像/动物)。(D)量化Col1α和Fn1基因转录的rt - pcr在肺部的车辆或nintedanib-treated TGFα老鼠(n= 4 /组)。所有数据都意味着±SEM。未配对学生的t以及用于测量组之间的统计学意义。*P< 0.05,* *P< 0.005。
确定的anti-fibrotic影响nintedanib也在一定程度上由于减少了扩散,我们执行ki - 67免疫染色测量总细胞增殖石蜡从TGFα小鼠肺部分处理nintedanib或车辆。nintedanib-treated老鼠ki - 67阳性细胞明显减少(图7 a, B)。经过分离和培养初级lung-resident从TGFα小鼠成纤维细胞对阿霉素2周nintedanib的存在与否,我们观察到剂量依赖性的影响在抑制nintedanib BrdU公司(图7 c)。此外,与细胞增殖相关的基因的表达,如CDK1,到,CCNA2,和PLK1显著减少lung-resident成纤维细胞的IPF患者nintedanib相比车辆(图7 d)。综合来看,这些研究都表明nintedanib阻止了生存,间充质细胞的增殖,ECM生产在活的有机体内。
图7
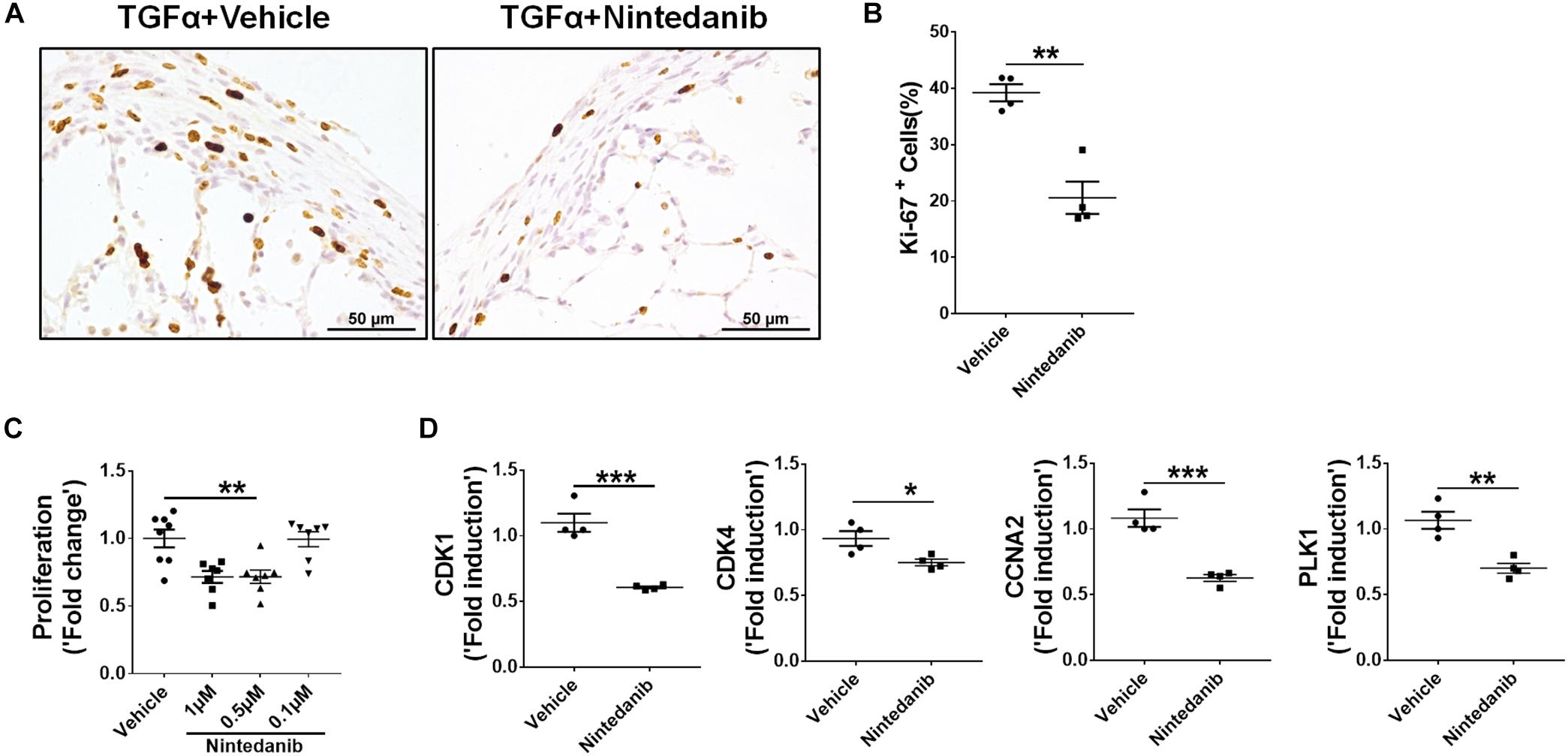
图7。期间Nintedanib变弱fibroproliferation TGFα-induced肺纤维化。(一)TGFα小鼠的肺组织化学染色部分处理车辆或nintedanib(60毫克/公斤)使用ki - 67 Ab。图像代表肺subpleura (n= 4 /组)。酒吧规模:50μm。(B)量化的百分比ki - 67+细胞subpleura地区从车辆或nintedanib-treated组(n= 4 /组;5代表图像/动物)。未配对学生的t以及用于测量组之间的统计学意义。(C)核扩散的量化使用BrdU合并分析在初级lung-resident TGFα小鼠成纤维细胞与基质细胞培养在强力霉素(阿霉素)为2周。与表示成纤维细胞治疗剂量的nintedanib总共48 h。叠化计算相对于vehicle-treated组。单向方差分析与Sidak多个比较测试被用来衡量一个显著差异。(D)CCNA2 CDK1量化,到,rt - pcr和PLK1基因转录的初级lung-resident人类成纤维细胞与IPF肺文化和处理车辆或nintedanib(1μM) 16 h (n= 4)。未配对的学生的t以及用于测量组之间的统计学意义。所有数据都意味着±SEM。*P< 0.05,* *P< 0.005,* * *P< 0.0005。
Nintedanib治疗变弱了肺纤维化的进展
确定长期nintedanib疗法减弱建立肝纤维化的进展,3周后强力霉素治疗纤维化时已经建立(Sontake et al ., 2017),TGFα老鼠nintedanib同时保持对阿霉素处理额外的4周(图8)。Non-TGFα和TGFα控制老鼠处理车辆同时保持对阿霉素的一个额外的4周。身体的重量TGFα老鼠处理车辆从基线下降到20%后7周的阿霉素(图8 b)。Nintedanib TGFα治疗小鼠第5周开始减速身体减肥相比vehicle-treated TGFα老鼠,剩下的身体重量低于车辆non-TGFα治疗控制老鼠。重要的是,肺总重量显著减毒TGFα小鼠接受nintedanib相比TGFα老鼠阿霉素和车辆治疗7周(图8 c)。我们观察到减少胶原蛋白沉积在肺部nintedanib相比汽车TGFα治疗治疗小鼠免疫印迹分析评估的胶原蛋白1在肺溶解产物α(图8 d肺部分)和马森的三色的染色(图8 e)。我们还观察到一个明显减少胸膜下适度调整周血管和支气管旁纤维化增厚与车辆相比,nintedanib治疗治疗TGFα老鼠(图8 f)。TGFα老鼠用强力霉素治疗发展严重纤维化肺力学的重大变化而non-TGFα控制老鼠阿霉素为7周。TGFαNintedanib治疗小鼠引发了降低肺阻力和倒电容和增加肺合规与车辆TGFα治疗小鼠相比图8 g)。流仪分析纤维细胞表现出绝对数量的减少TGFα老鼠的肺纤维细胞人口管理与nintedanib相比,车辆(图8 h和表3)。综合来看,这些研究都表明nintedanib疗法建立肝纤维化时调节纤维化疾病的恶化TGFα老鼠基于生理和组织学参数。
表3

表3。流仪绝对数量的纤维细胞的分析。
图8
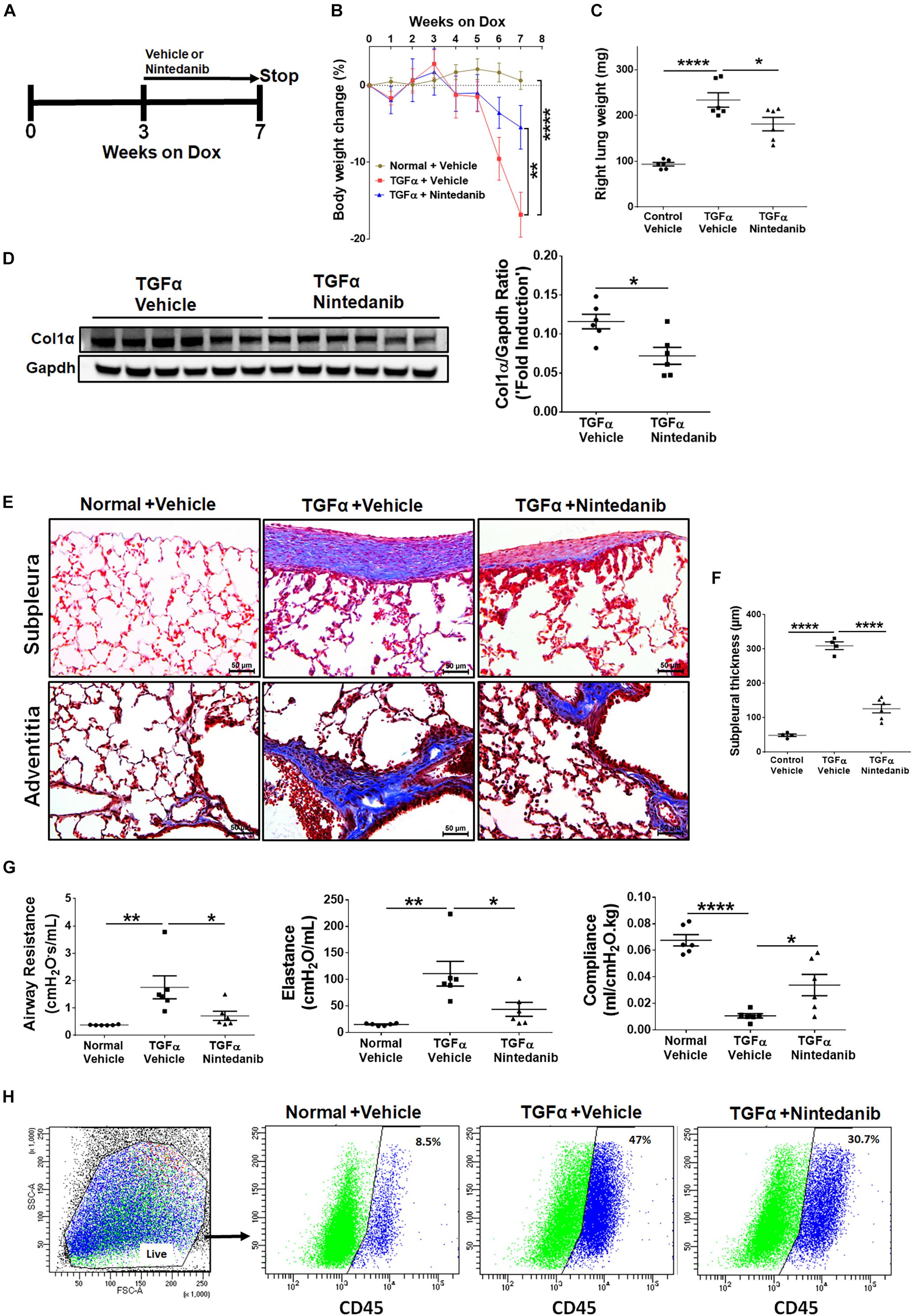
图8。Nintedanib治疗变弱建立和持续的肺纤维化在活的有机体内。(一)的示意图表示nintedanib治疗协议。控制老鼠和TGFα老鼠放在阿霉素3周然后处理车辆或nintedanib(60毫克/公斤;一天一次)4周而继续强力霉素总共7周(n= 6 /组)。(B)小鼠的体重变化百分比车辆和nintedanib对待。(C)定量所有组对小鼠的肺重量的车辆和nintedanib对待。(D)定量免疫印迹肺胶原蛋白1α的溶解产物由TGFα老鼠处理车辆或nintedanib (n= 6 /组)。(E)马森的三色的彩色图像肺部分的组。前肺,胸膜下区;底动脉外膜。酒吧,规模50μm。图像是每组的代表(n= 5 - 6 /组)。(F)胸膜下肺部分的厚度测量老鼠沾着马森的三色的。(G)所有组的肺功能变化的老鼠车辆和nintedanib对待。(H)代表流仪分析情节展示CD45 + ve纤维细胞百分比的总肺间质细胞与车辆和nintedanib小鼠。统计学意义是用单向ANNOVA Sidak的多重比较检验。*p< 0.05,* *p< 0.005,* * * *p< 0.00005。
讨论
IPF是一种进行性纤维化肺病的特征是持续积累各种间充质细胞,产生过量的ECM形成不可逆转的肺部疤痕组织。基于转录组分析的生存途径,我们建议IPF和TGFα模型调解的发展在纤维细胞凋亡抵抗和myofibroblasts很大程度上类似的机制导致纤维化病变在肺纤维化发病机制的不断扩大。
纤维细胞升高在几个慢性肺部疾病,肺纤维化病变包括IPF (Moeller et al ., 2009)。我们之前的研究结果表明,它们积聚在肺部的病变而不是普通区域在TGFα-induced肺纤维化(Madala et al ., 2014 b)。我们的新结果强烈支持这个想法,纤维细胞(CD45+Col1+)积聚在纤维化病变,因为他们成为抵抗细胞凋亡。证明纤维细胞和lung-resident myofibroblasts pro-apoptotic低水平表达的蛋白质,包括贝克和伯灵顿,我们的研究结果支持他们产生耐药性Fas-mediated凋亡的前提,进一步证明了这一点他们降低裂解caspase-3/7 Fas-activating抗体处理时激活。越来越多的证据表明,纤维细胞的直接贡献myofibroblast池是有限的,但它们分泌几个lung-resident成纤维细胞的旁分泌因子诱导激活;具体来说,fibroproliferation和myofibroblast转换在肺纤维化的发病机制(桥本et al ., 2004;Madala et al ., 2014 b;Sontake et al ., 2015;阿什利et al ., 2017)。因此,针对lung-resident成纤维细胞和纤维细胞有必要寻找策略来减弱肺纤维化。
在这里,我们使用比较表达式分析识别apoptosis-linked基因,包括midikine gremlin1, Spp1, Ptgs2,贝克,伯灵顿,Bcl2, Fas, IPF TGFα-induced肺纤维化小鼠模型,通过rt - pcr验证表达式的变化,发现一个明显的重叠。在调节基因,midikine、Spp1 gremlin1被证明函数作为积极的IPF的监管机构和在bleomycin-induced肺纤维化(Pardo et al ., 2005;Koli et al ., 2006;对剧中et al ., 2017)。使用癌症细胞系的研究进一步支持了假设增加这些基因的表达间充质细胞的存活率增加,表明其抑制作为干预策略(Tamminen et al ., 2013;穆勒et al ., 2014;萨利赫et al ., 2016;湘et al ., 2017)。特别是,抑制gremlin-1足以诱导的表达paclitaxel-mediated间皮瘤肿瘤细胞系凋亡(Tamminen et al ., 2013)。小鬼1还能抑制BMP4,已经被证明可以诱导细胞凋亡在角膜成纤维细胞(Mohan et al ., 1998)。
支持和抗凋亡的监管机构之间的平衡可能决定细胞命运。在这里,我们的数据表明,几个pro-apoptotic基因转录,包括贝克,伯灵顿,和Fas表达下调IPF的肺纤维化和TGFα老鼠。注意,贝克和伯灵顿在纤维细胞表达下调和myofibroblasts隔绝TGFα小鼠的肺。在支持,小鼠胚胎成纤维细胞的生存(mef)隔绝伯灵顿和Bak-null老鼠在bleomycin-induced增强细胞死亡(李et al ., 2005)。同样,Fas受体的转录水平,提升者外在凋亡细胞死亡在Fas配体活化的小鼠肺纤维化的含量很低,与先前的研究一致的(多迪et al ., 2018)。此外,我们观察到抵抗细胞死亡Fas-mediated纤维细胞和lung-resident myofibroblasts。总的来说,我们的研究结果表明内在和外在的参与凋亡通路的监管机构间充质细胞生存在肺纤维化的发病机制。未来的研究使用转基因操作和过继转移或纤维细胞的损耗将识别的分子监管机构生存和他们的角色在肺纤维化的不同阶段。
尽管先前的研究发现anti-fibrotic nintedanib多个模型的肺纤维化的影响,我们观察到不同的影响衰减建立和持续的肺纤维化的nintedanib TGFα老鼠。特别是,我们表明,nintedanib纤维细胞凋亡增加间隙和lung-resident myofibroblasts减弱TGFα-induced肺纤维化的进展。在最近的一项研究中,nintedanib显示诱导自噬在肺成纤维细胞与IPF (Rangarajan周二et al ., 2016)。此外,nintedanib可以抑制纤维细胞迁移,从而减少纤维细胞的数量在肺bleomycin-induced肺纤维化(佐藤et al ., 2017)。与先前的报道一致,我们的
在体外和在活的有机体内研究证实,nintedanib治疗变弱扩散和proliferation-associated基因的表达。我们的新发现表明,nintedanib抑制纤维细胞的生存和lung-resident myofibroblasts诱导pro-apoptotic基因表达在严重纤维化肺病。然而,上述影响可能并不局限于间充质细胞和其他肺细胞如上皮细胞易受nintedanib,这可能有益或有害影响正常的再生过程在肺部受伤。在最近的一项研究支持莱曼et al。(2018)表明nintedanib治疗稳定远端上皮细胞标记物的表达,比如SP-C小鼠和人肺组织的文化。因此,未来的研究是必要的理解nintedanib在多个肺细胞的剂量依赖效应包括间叶细胞、上皮细胞、内皮细胞和免疫细胞参与anti-fibrotic和修复过程。这些研究将是关键的开发更有效的治疗对严重纤维化肺病。然而,临床开发有效的联合疗法可以反向建立和持续的纤维化受到缺乏知识的作用机制,也不受欢迎的副作用与nintedanib治疗有关。调节成纤维细胞的活性可能作为一种有价值的方法来治疗严重的肺纤维化疾病,特别是多种生长因子的活性。我们先前的研究证明多种生长因子包括TGFβupregulation, il - 6, IL-13, amphiregulin和epiregulin截然不同的间充质细胞子集如纤维细胞、肺居民成纤维细胞和WT1-positive myofibroblasts TGFα-induced肺纤维化(Madala et al ., 2014 c;Sontake et al ., 2015,2018年)。TGFα发现调节在多个肺纤维化疾病包括IPF和囊性纤维化(鲍曼et al ., 1999;艰苦的et al ., 1999)。一些报告表明,抑制多个信号通路显著防止TGFα-induced肺纤维化(Madala et al ., 2014 a,2016年,一个,b)。然而,完整的解决肺纤维化与nintedanib疗法和其他抑制剂TGFα模型并不是唯一的。在最近的一项研究中使用热议小鼠关节疾病Redente et al。(2018)发现nintedanib治疗变弱胶原沉积,但没有改变肺合规。nintedanib的变异性的影响可能反映差异的相对贡献多个pro-fibrotic在调节纤维发生蛋白质和途径在小鼠的肝纤维化模型和压力进行了研究。
总之,我们发现了一个抗细胞凋亡的作用在调节纤维细胞积累在肺纤维化的发病机制。我们的新结果表明nintedanib肺纤维细胞和凋亡居民myofibroblasts。凋亡的增加间隙pro-apoptotic基因的表达增加有关贝克和伯灵顿nintedanib (图9)。我们的在体外研究中使用的主要细胞在活的有机体内研究使用鼠标TGFα-induced肺纤维化模型表明nintedanib疗法是有效的减弱成纤维细胞激活包括fibroproliferation、抗凋亡和ECM生产成熟的肺纤维化病变。我们的新发现强调使用鼠标的相关性TGFα-induced肺纤维化模型,来研究成纤维细胞激活肺纤维化疾病的发病机理。未来的研究运用这个模型单独或结合其他慢性肺纤维化疾病表型不解决将有用的检查肺再生的操作和测试的有效性nintedanib IPF结合其他药物。
图9
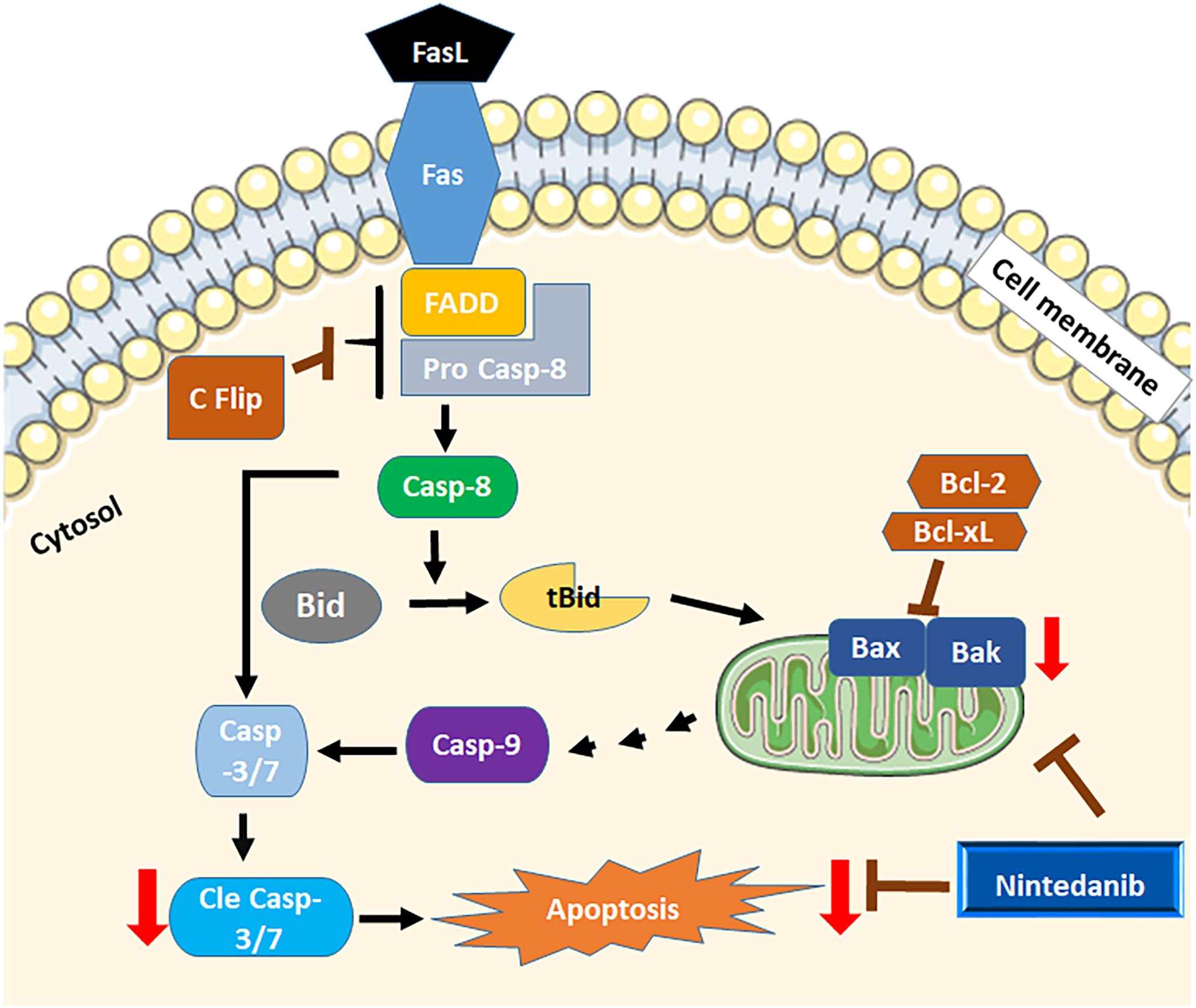
图9。Schematicview nintedanib的细胞凋亡。内在和外在凋亡监管机构间充质细胞中特异表达的肺纤维化。Nintedanib凋亡增加pro-apoptotic Bak基因的表达和伯灵顿。
数据可用性
公开的数据集进行分析。这些数据可以在这里找到:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE53845。
道德声明
本研究按照协议进行了机构的动物保健和使用委员会批准的辛辛那提儿童医院的研究基础。
作者的贡献
RK和SM的构思和设计研究。RK, SM, AJ, GR进行实验和写的手稿。
资金
这项工作是由国家卫生研究院(1 r01 HL134801 1一下r21 AI137309, w81xwh - 17 - 1 - 0666) (SM)、卫生部门研究,印度政府(GR)和生物技术(印度生物技术部)奖学金,印度政府(RK)。
免责声明
所有的作者都有一个金融与商业实体的关系感兴趣的主题这手稿。
利益冲突声明
作者声明,这项研究是在没有进行任何商业或财务关系可能被视为一个潜在的利益冲突。
确认
作者感谢兽医服务和病理学研究核心辛辛那提儿童医院医学中心的帮助在这个研究。
引用
Ajayi, i O。,Sisson, T. H., Higgins, P. D., Booth, A. J., Sagana, R. L., Huang, S. K., et al. (2013). X-linked inhibitor of apoptosis regulates lung fibroblast resistance to Fas-mediated apoptosis.点。j .和。细胞摩尔。生物。49岁,86 - 95。doi: 10.1165 / rcmb.2012 - 0224摄氏度
希礼,s . L。,Wilke, C. A., Kim, K. K., and Moore, B. B. (2017). Periostin regulates fibrocyte function to promote myofibroblast differentiation and lung fibrosis.粘膜Immunol。10日,341 - 351。doi: 10.1038 / mi.2016.61
巴雷特,T。,Troup, D. B., Wilhite, S. E., Ledoux, P., Rudnev, D., Evangelista, C., et al. (2007). NCBI GEO: mining tens of millions of expression profiles–database and tools update.核酸Res。35岁,D760-D765。doi: 10.1093 / nar / gkl887
记述,r . P。低,E·E。,Miller, M. A., Bejarano, P. A., and Heffelfinger, S. C. (1999). Overexpression of transforming growth factor-alpha and epidermal growth factor-receptor in idiopathic pulmonary fibrosis.结节病Vasc。弥漫性肺说。16日,57 - 61。
陈,J。,Bardes, E. E., Aronow, B. J., and Jegga, A. G. (2009). ToppGene suite for gene list enrichment analysis and candidate gene prioritization.核酸Res。37岁的W305-W311。doi: 10.1093 / nar / gkp427
DePianto d J。,Chandriani, S., Abbas, A. R., Jia, G., N’Diaye, E. N., Caplazi, P., et al. (2015). Heterogeneous gene expression signatures correspond to distinct lung pathologies and biomarkers of disease severity in idiopathic pulmonary fibrosis.胸腔70年,48-56。doi: 10.1136 / thoraxjnl - 2013 - 204596
Desmouliere,。Redard, M。Darby,我。,和Gabbiani, G. (1995). Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar.点。j .分册。146年,56 - 66。
多迪,a E。,Ajayi, i O。常,C。,胡子,M。,希礼,s . L。,黄,美国K。,et al. (2018). Regulation of fibroblast Fas expression by soluble and mechanical pro-fibrotic stimuli.和。Res。19:91。doi: 10.1186 / s12931 - 018 - 0801 - 4
弗兰克尔,美国K。,Cosgrove, G. P., Cha, S. I., Cool, C. D., Wynes, M. W., Edelman, B. L., et al. (2006). TNF-alpha sensitizes normal and fibrotic human lung fibroblasts to Fas-induced apoptosis.点。j .和。细胞摩尔。生物。34岁,293 - 304。doi: 10.1165 / rcmb.2005 - 0155摄氏度
弗里曼·m·R。Sathish, V。Manlove, L。王,S。,Britt, R. D., Thompson, M. A., et al. (2017). Brain-derived neurotrophic factor and airway fibrosis in asthma.点。j .杂志。肺细胞。摩尔。杂志。313年,L360-L370。doi: 10.1152 / ajplung.00580.2016
格拉瑟,s W。,Hagood, J. S., Wong, S., Taype, C. A., Madala, S. K., and Hardie, W. D. (2016). Mechanisms of lung fibrosis resolution.点。j .分册。186年,1066 - 1077。doi: 10.1016 / j.ajpath.2016.01.018
Golan-Gerstl, R。,Wallach-Dayan, S. B., Zisman, P., Cardoso, W. V., Goldstein, R. H., and Breuer, R. (2012). Cellular FLICE-like inhibitory protein deviates myofibroblast fas-induced apoptosis toward proliferation during lung fibrosis.点。j .和。细胞摩尔。生物。47岁,271 - 279。doi: 10.1165 / rcmb.2010 - 0284 rc
格里宾,J。,Hubbard, R. B., Le Jeune, I., Smith, C. J., West, J., and Tata, L. J. (2006). Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK.胸腔61年,980 - 985。doi: 10.1136 / thx.2006.062836
难的,w . D。,Bejarano, P. A., Miller, M. A., Yankaskas, J. R., Ritter, J. H., Whitsett, J. A., et al. (1999). Immunolocalization of transforming growth factor alpha and epidermal growth factor receptor in lungs of patients with cystic fibrosis.Pediatr。Dev,病理学研究。2,415 - 423。doi: 10.1007 / s100249900144
难的,w . D。,格拉瑟,s W。,和Hagood, J. S. (2009). Emerging concepts in the pathogenesis of lung fibrosis.点。j .分册。175年,3-16。doi: 10.2353 / ajpath.2009.081170
难的,w . D。,Le Cras, T. D., Jiang, K., Tichelaar, J. W., Azhar, M., and Korfhagen, T. R. (2004). Conditional expression of transforming growth factor-alpha in adult mouse lung causes pulmonary fibrosis.点。j .杂志。肺细胞。摩尔。杂志。286年,L741-L749。doi: 10.1152 / ajplung.00208.2003
桥本,N。金,H。刘,T。,Chensue, S. W., and Phan, S. H. (2004). Bone marrow-derived progenitor cells in pulmonary fibrosis.j .中国。投资。113年,243 - 252。doi: 10.1172 / JCI18847
Hilberg F。,Roth, G. J., Krssak, M., Kautschitsch, S., Sommergruber, W., Tontsch-Grunt, U., et al. (2008). BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy.癌症Res。68年,4774 - 4782。doi: 10.1158 / 0008 - 5472. -能- 07 - 6307
霍洛维茨,j . C。、Ajayi i O。,Kulasekaran, P., Rogers, D. S., White, J. B., Townsend, S. K., et al. (2012). Survivin expression induced by endothelin-1 promotes myofibroblast resistance to apoptosis.学生物化学Int。j。细胞生物。44岁,158 - 169。doi: 10.1016 / j.biocel.2011.10.011
黄,J。,Beyer, C., Palumbo-Zerr, K., Zhang, Y., Ramming, A., Distler, A., et al. (2016). Nintedanib inhibits fibroblast activation and ameliorates fibrosis in preclinical models of systemic sclerosis.安。感冒。说。75年,883 - 890。doi: 10.1136 / annrheumdis - 2014 - 207109
黄,美国K。,和霍洛维茨,j . C。(2014). Outstaying their welcome: the persistent myofibroblast in IPF.奥斯汀j . Pulm。和。地中海。1:3。
哈钦森,j . P。,McKeever t M。福格蒂,a W。Navaratnam, V。,和Hubbard, R. B. (2014). Increasing global mortality from idiopathic pulmonary fibrosis in the twenty-first century.安。点。Thorac。Soc。11日,1176 - 1185。doi: 10.1513 / annalsats.201404 - 145摄氏度
国王,t·E。,Bradford, W. Z., Castro-Bernardini, S., Fagan, E. A., Glaspole, I., Glassberg, M. K., et al. (2014). A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis.心血管病。j .地中海。370年,2083 - 2092。doi: 10.1056 / NEJMoa1402582
Kleaveland, k . R。,摩尔,B, B。,和Kim, K. K. (2014). Paracrine functions of fibrocytes to promote lung fibrosis.启和专家。地中海。8,163 - 172。doi: 10.1586 / 17476348.2014.862154
Koli, K。,Myllarniemi, M., Vuorinen, K., Salmenkivi, K., Ryynanen, M. J., Kinnula, V. L., et al. (2006). Bone morphogenetic protein-4 inhibitor gremlin is overexpressed in idiopathic pulmonary fibrosis.点。j .分册。169年,61 - 71。doi: 10.2353 / ajpath.2006.051263
李,诉Y。,Schroedl, C., Brunelle, J. K., Buccellato, L. J., Akinci, O. I., Kaneto, H., et al. (2005). Bleomycin induces alveolar epithelial cell death through JNK-dependent activation of the mitochondrial death pathway.点。j .杂志。肺细胞。摩尔。杂志。289年,L521-L528。doi: 10.1152 / ajplung.00340.2004
莱曼,M。镶嵌细工,L。,Alsafadi, H. N., Klee, S., Hermann, S., Mutze, K., et al. (2018). Differential effects of nintedanib and pirfenidone on lung alveolar epithelial cell function in ex vivo murine and human lung tissue cultures of pulmonary fibrosis.和。Res。19:175。doi: 10.1186 / s12931 - 018 - 0876 - y
李,l F。,Kao, K. C., Liu, Y. Y., Lin, C. W., Chen, N. H., Lee, C. S., et al. (2017). Nintedanib reduces ventilation-augmented bleomycin-induced epithelial-mesenchymal transition and lung fibrosis through suppression of the Src pathway.地中海摩尔。j .细胞。21日,2937 - 2949。doi: 10.1111 / jcmm.13206
Madala美国K。,Edukulla, R., Davis, K. R., Schmidt, S., Davidson, C., Kitzmiller, J. A., et al. (2012). Resistin-like molecule alpha1 (Fizz1) recruits lung dendritic cells without causing pulmonary fibrosis.和。Res。接下来。doi: 10.1186 / 1465-9921-13-51
Madala美国K。,Edukulla, R., Phatak, M., Schmidt, S., Davidson, C., Acciani, T. H., et al. (2014a). Dual targeting of MEK and PI3K pathways attenuates established and progressive pulmonary fibrosis.《公共科学图书馆•综合》9:e86536。doi: 10.1371 / journal.pone.0086536
Madala美国K。,Edukulla, R., Schmidt, S., Davidson, C., Ikegami, M., and Hardie, W. D. (2014b). Bone marrow-derived stromal cells are invasive and hyperproliferative and alter transforming growth factor-alpha-induced pulmonary fibrosis.点。j .和。细胞摩尔。生物。50岁,777 - 786。doi: 10.1165 / rcmb.2013 - 0042摄氏度
Madala美国K。,Korfhagen, T. R., Schmidt, S., Davidson, C., Edukulla, R., Ikegami, M., et al. (2014c). Inhibition of the alphavbeta6 integrin leads to limited alteration of TGF-alpha-induced pulmonary fibrosis.点。j .杂志。肺。细胞。摩尔。杂志。306年,L726-L735。doi: 10.1152 / ajplung.00357.2013
Madala美国K。Sontake, V。,Edukulla, R., Davidson, C. R., Schmidt, S., and Hardie, W. D. (2016a). Unique and redundant functions of p70 ribosomal S6 kinase isoforms regulate mesenchymal cell proliferation and migration in pulmonary fibrosis.点。j .和。细胞摩尔。生物。55岁,792 - 803。doi: 10.1165 / rcmb.2016 - 0090摄氏度
Madala美国K。托马斯·G。,Edukulla, R., Davidson, C., Schmidt, S., Schehr, A., et al. (2016b). p70 ribosomal S6 kinase regulates subpleural fibrosis following transforming growth factor-alpha expression in the lung.点。j .杂志。肺细胞。摩尔。杂志。310年,L175-L186。doi: 10.1152 / ajplung.00063.2015
大师,S。,Shimbori, C., and Kolb, M. (2013). Fibrocytes in pulmonary fibrosis: a brief synopsis.欧元。和。牧师。22日,552 - 557。doi: 10.1183/09059180.00007713
Marudamuthu, a S。Shetty,美国K。,Bhandary, Y. P., Karandashova, S., Thompson, M., Sathish, V., et al. (2015). Plasminogen activator inhibitor-1 suppresses profibrotic responses in fibroblasts from fibrotic lungs.生物。化学。290年,9428 - 9441。doi: 10.1074 / jbc.M114.601815
对剧中,K。,Tanino, Y., Wang, X., Nikaido, T., Kikuchi, M., Sato, Y., et al. (2017). Involvement of midkine in the development of pulmonary fibrosis.杂志。代表。5:e13383。doi: 10.14814 / phy2.13383
Moeller,。,Gilpin, S. E., Ask, K., Cox, G., Cook, D., Gauldie, J., et al. (2009). Circulating fibrocytes are an indicator of poor prognosis in idiopathic pulmonary fibrosis.点。j .和。暴击。保健医疗。179年,588 - 594。doi: 10.1164 / rccm.200810 - 1534摄氏度
莫汉,R R。金,w·J。,莫汉,R R。陈,L。,和Wilson, S. E. (1998). Bone morphogenic proteins 2 and 4 and their receptors in the adult human cornea.投资。角膜切削。粘度科学。39岁,2626 - 2636。
摩尔,B, B。,Kolodsick, J. E., Thannickal, V. J., Cooke, K., Moore, T. A., Hogaboam, C., et al. (2005). CCR2-mediated recruitment of fibrocytes to the alveolar space after fibrotic injury.点。j .分册。166年,675 - 684。doi: 10.1016 / s0002 - 9440 (10) 62289 - 4
摩尔,B, B。穆雷,L。Das,。,Wilke, C. A., Herrygers, A. B., and Toews, G. B. (2006). The role of CCL12 in the recruitment of fibrocytes and lung fibrosis.点。j .和。细胞摩尔。生物。35岁,175 - 181。doi: 10.1165 / rcmb.2005 - 0239摄氏度
穆勒,我。。,Chatterjee, M., Schneider, M., Borst, O., Seizer, P., Schonberger, T., et al. (2014). Gremlin-1 inhibits macrophage migration inhibitory factor-dependent monocyte function and survival.Int。j .心功能杂志。176年,923 - 929。doi: 10.1016 / j.ijcard.2014.08.051
内森,s D。Albera C。,Bradford, W. Z., Costabel, U., Glaspole, I., Glassberg, M. K., et al. (2017). Effect of pirfenidone on mortality: pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis.和出版的《柳叶刀》杂志上。地中海。5,33-41。doi: 10.1016 / s2213 - 2600 (16) 30326 - 5
Nho, r S。,Peterson, M., Hergert, P., and Henke, C. A. (2013). FoxO3a (Forkhead Box O3a) deficiency protects idiopathic pulmonary fibrosis (IPF) fibroblasts from type I polymerized collagen matrix-induced apoptosis via caveolin-1 (cav-1) and Fas.《公共科学图书馆•综合》8:e61017。doi: 10.1371 / journal.pone.0061017
Pardo。,Gibson, K., Cisneros, J., Richards, T. J., Yang, Y., Becerril, C., et al. (2005). Up-regulation and profibrotic role of osteopontin in human idiopathic pulmonary fibrosis.科学硕士。2:e251。doi: 10.1371 / journal.pmed.0020251
抗起球,D。、风扇、T。,Huang, D., Kaul, B., and Gomer, R. H. (2009). Identification of markers that distinguish monocyte-derived fibrocytes from monocytes, macrophages, and fibroblasts.《公共科学图书馆•综合》4:e7475。doi: 10.1371 / journal.pone.0007475
抗起球,D。,和Gomer, R. H. (2014). Persistent lung inflammation and fibrosis in serum amyloid P component (APCs-/-) knockout mice.《公共科学图书馆•综合》9:e93730。doi: 10.1371 / journal.pone.0093730
抗起球,D。Roife D。王,M。,Ronkainen, S. D., Crawford, J. R., Travis, E. L., et al. (2007). Reduction of bleomycin-induced pulmonary fibrosis by serum amyloid P.j . Immunol。179年,4035 - 4044。doi: 10.4049 / jimmunol.179.6.4035
贾拉。,Kurundkar, A., Kurundkar, D., Bernard, K., Sanders, Y. Y., Ding, Q., et al. (2016). novel mechanisms for the antifibrotic action of nintedanib.点。j .和。细胞摩尔。生物。54岁,51-59。doi: 10.1165 / rcmb.2014 - 0445摄氏度
Redente, e . F。,Aguilar, M. A., Black, B. P., Edelman, B. L., Bahadur, A. N., Humphries, S. M., et al. (2018). Nintedanib reduces pulmonary fibrosis in a model of rheumatoid arthritis-associated interstitial lung disease.点。j .杂志。肺细胞。摩尔。杂志。314年,L998-L1009。doi: 10.1152 / ajplung.00304.2017
Richeldi, L。,du Bois, R. M., Raghu, G., Azuma, A., Brown, K. K., Costabel, U., et al. (2014). Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis.心血管病。j .地中海。370年,2071 - 2082。doi: 10.1056 / NEJMoa1402584
Safaeian, L。在床上,。,和Vaseghi, G. (2014). The role of Bcl-2 family proteins in pulmonary fibrosis.欧元。j .杂志。741年,281 - 289。doi: 10.1016 / j.ejphar.2014.07.029
萨利赫。,Thompson, D. E., McConkey, J., Murray, P., and Moorehead, R. A. (2016). Osteopontin regulates proliferation, apoptosis, and migration of murine claudin-low mammary tumor cells.BMC癌症16:359。doi: 10.1186 / s12885 - 016 - 2396 - 9
佐藤,S。,Shinohara, S., Hayashi, S., Morizumi, S., Abe, S., Okazaki, H., et al. (2017). Anti-fibrotic efficacy of nintedanib in pulmonary fibrosis via the inhibition of fibrocyte activity.和。Res。18:172。doi: 10.1186 / s12931 - 017 - 0654 - 2
香农,P。,Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks.基因组Res。13日,2498 - 2504。doi: 10.1101 / gr.1239303
辛格,B。,Kasam, R. K., Sontake, V., Wynn, T. A., and Madala, S. K. (2017). Repetitive intradermal bleomycin injections evoke T-helper cell 2 cytokine-driven pulmonary fibrosis.点。j .杂志。肺细胞。摩尔。杂志。313年,L796-L806。doi: 10.1152 / ajplung.00184.2017
Sontake, V。,Gajjala, P. R., Kasam, R. K., and Madala, S. K. (2019). New therapeutics based on emerging concepts in pulmonary fibrosis.当今专家。其他。目标23日,69 - 81。doi: 10.1080 / 14728222.2019.1552262
Sontake, V。,Kasam, R. K., Sinner, D., Korfhagen, T. R., Reddy, G. B., White, E. S., et al. (2018). Wilms’ tumor 1 drives fibroproliferation and myofibroblast transformation in severe fibrotic lung disease.江森自控的洞察力3:121252。doi: 10.1172 / jci.insight.121252
Sontake, V。,Shanmukhappa, S. K., DiPasquale, B. A., Reddy, G. B., Medvedovic, M., Hardie, W. D., et al. (2015). Fibrocytes regulate wilms tumor 1-Positive cell accumulation in severe fibrotic lung disease.j . Immunol。195年,3978 - 3991。doi: 10.4049 / jimmunol.1500963
Sontake, V。王,Y。,Kasam, R. K., Sinner, D., Reddy, G. B., Naren, A. P., et al. (2017). Hsp90 regulation of fibroblast activation in pulmonary fibrosis.江森自控的洞察力2:e91454。doi: 10.1172 / jci.insight.91454
Tamminen, j . A。,Parviainen, V., Ronty, M., Wohl, A. P., Murray, L., Joenvaara, S., et al. (2013). Gremlin-1 associates with fibrillin microfibrils in vivo and regulates mesothelioma cell survival through transcription factor slug.肿瘤形成2:e66。doi: 10.1038 / oncsis.2013.29
田中,T。,Yoshimi, M., Maeyama, T., Hagimoto, N., Kuwano, K., and Hara, N. (2002). Resistance to Fas-mediated apoptosis in human lung fibroblast.欧元。和。J。20岁,359 - 368。doi: 10.1183 / 09031936.02.00252602
Thannickal诉J。,和霍洛维茨,j . C。(2006). Evolving concepts of apoptosis in idiopathic pulmonary fibrosis.Proc。。Thorac。Soc。3,350 - 356。doi: 10.1513 / pats.200601 - 001 tk
Wollin, L。,Maillet, I., Quesniaux, V., Holweg, A., and Ryffel, B. (2014). Antifibrotic and anti-inflammatory activity of the tyrosine kinase inhibitor nintedanib in experimental models of lung fibrosis.j .杂志。其他实验。349年,209 - 220。doi: 10.1124 / jpet.113.208223
Wollin, L。Wex E。,Pautsch, A., Schnapp, G., Hostettler, K. E., Stowasser, S., et al. (2015). Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis.欧元。和。J。45岁,1434 - 1445。doi: 10.1183/09031936.00174914
关键字:myofibroblasts,纤维细胞,细胞凋亡,特发性肺纤维化,nintedanib
引用:Kasam RK, Reddy GB, Jegga AG)和Madala SK(2019)间充质细胞生存通路的失调严重纤维化肺病:Nintedanib疗法的效果。前面。杂志。10:532。doi: 10.3389 / fphar.2019.00532
收到:2019年2月24日;接受:2019年4月29日;
发表:2019年5月17日。
编辑:
Narasaiah Kolliputi美国南佛罗里达大学版权Reddy,©2019 Kasam Jegga Madala。这是一个开放分布式根据文章知识共享归属许可(CC)。使用、分发或复制在其他论坛是允许的,提供了原始作者(年代)和著作权人(s)认为,最初发表在这个期刊引用,按照公认的学术实践。没有使用、分发或复制是不符合这些条件的允许。
*通信:Satish k . Madalasatish.madala@cchmc.org